As confidentially submitted to the Securities and Exchange Commission on December 23, 2025. This draft registration statement has not been publicly filed with the Securities and Exchange Commission and all information herein remains strictly confidential.
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Generate Biomedicines, Inc.
(Exact name of registrant as specified in its charter)
Delaware |
2834 |
83-1630228 |
(State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
101 South Street, Suite 900
Somerville, MA 02143
(888) 469-0055
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Michael Nally
President and Chief Executive Officer
Generate Biomedicines, Inc.
101 South Street, Suite 900
Somerville, MA 02143
(888) 469-0055
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Stuart M. Cable Joseph C. Theis Stephanie Richards Janet Hsueh Goodwin Procter LLP 100 Northern Avenue Boston, MA 02210 (617) 570-1000 |
Sean Martin Chief Legal Officer and General Counsel Generate Biomedicines, Inc. 101 South Street, Suite 900 Somerville, MA 02143 (888) 469-0055 |
Peter N. Handrinos Wesley C. Holmes Latham & Watkins LLP 200 Clarendon Street Boston, MA 02116 (617) 880-4500 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: ☐
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
☐ |
Accelerated filer |
☐ |
Non-accelerated filer |
☒ |
Smaller reporting company |
☒ |
|
|
Emerging growth company |
☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.


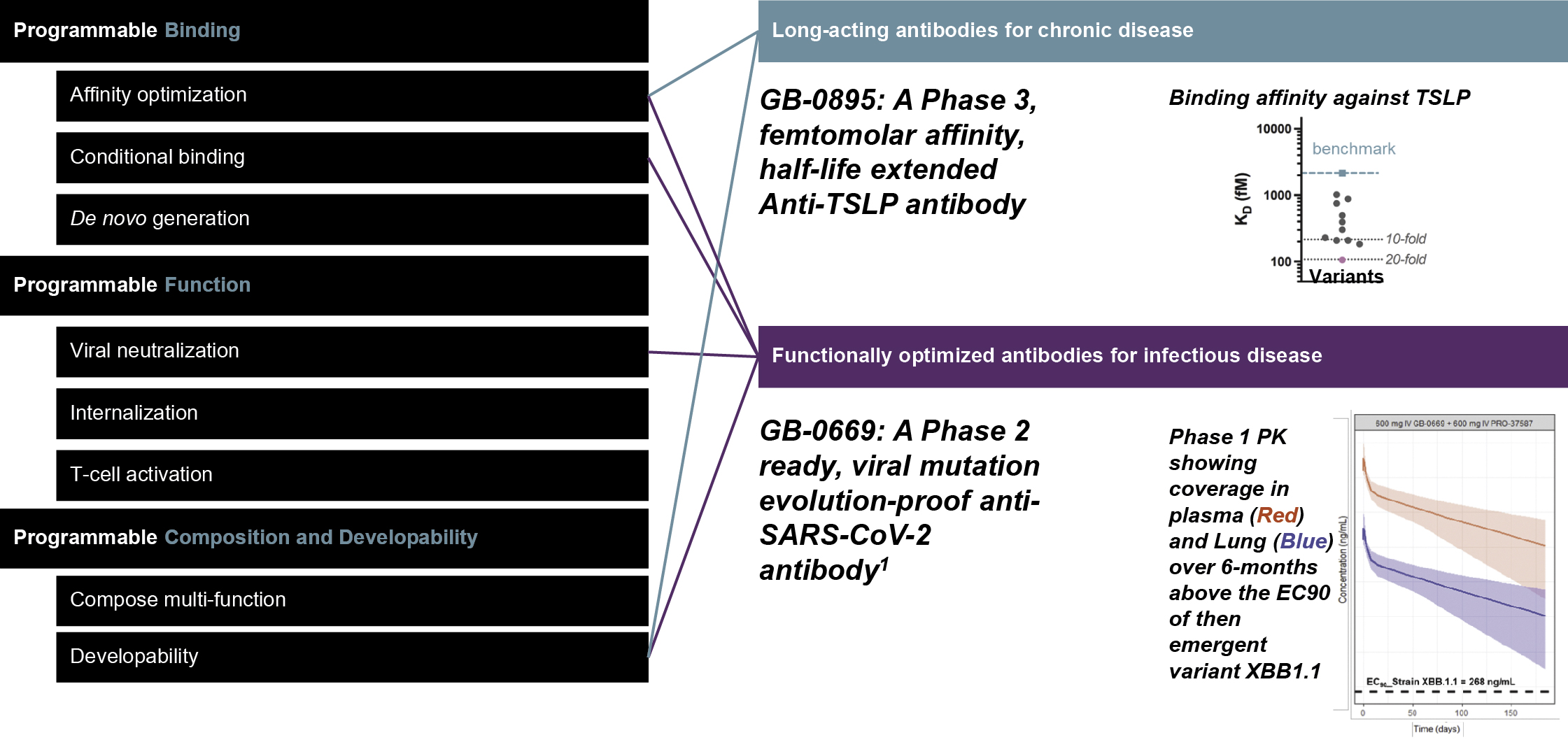
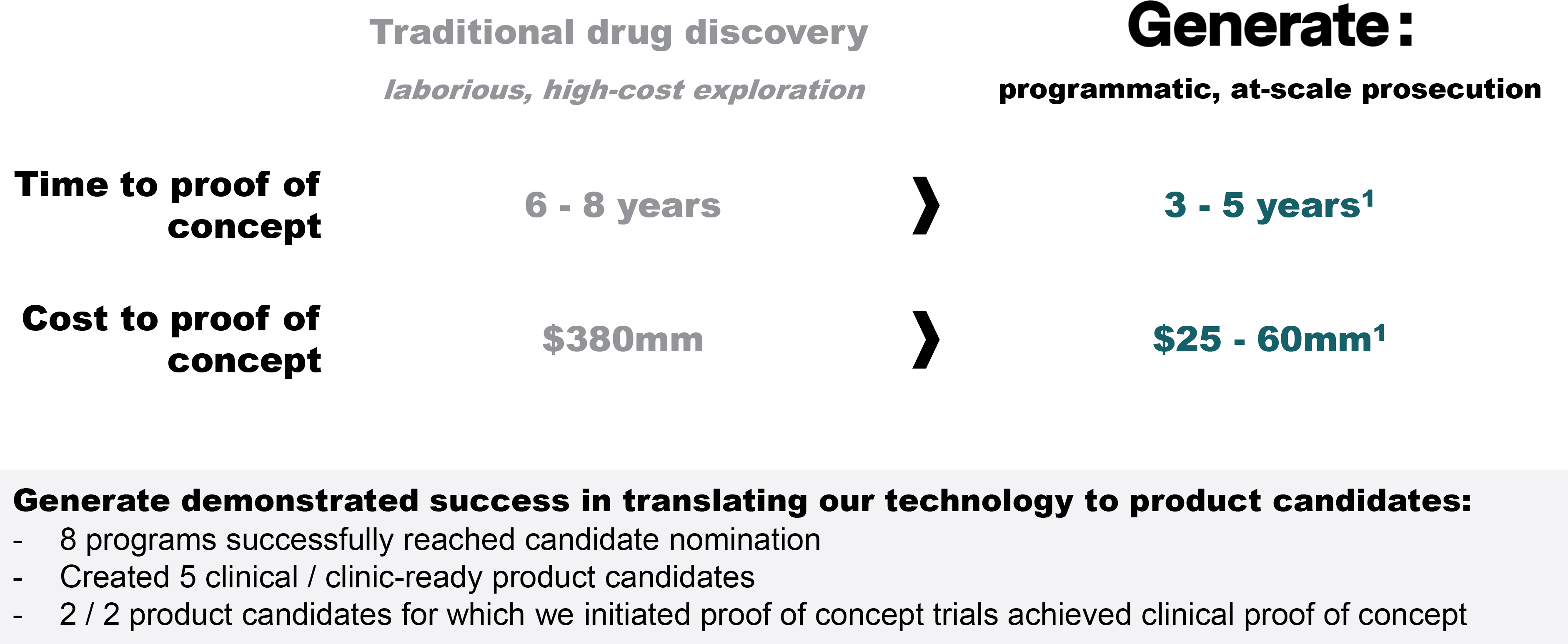
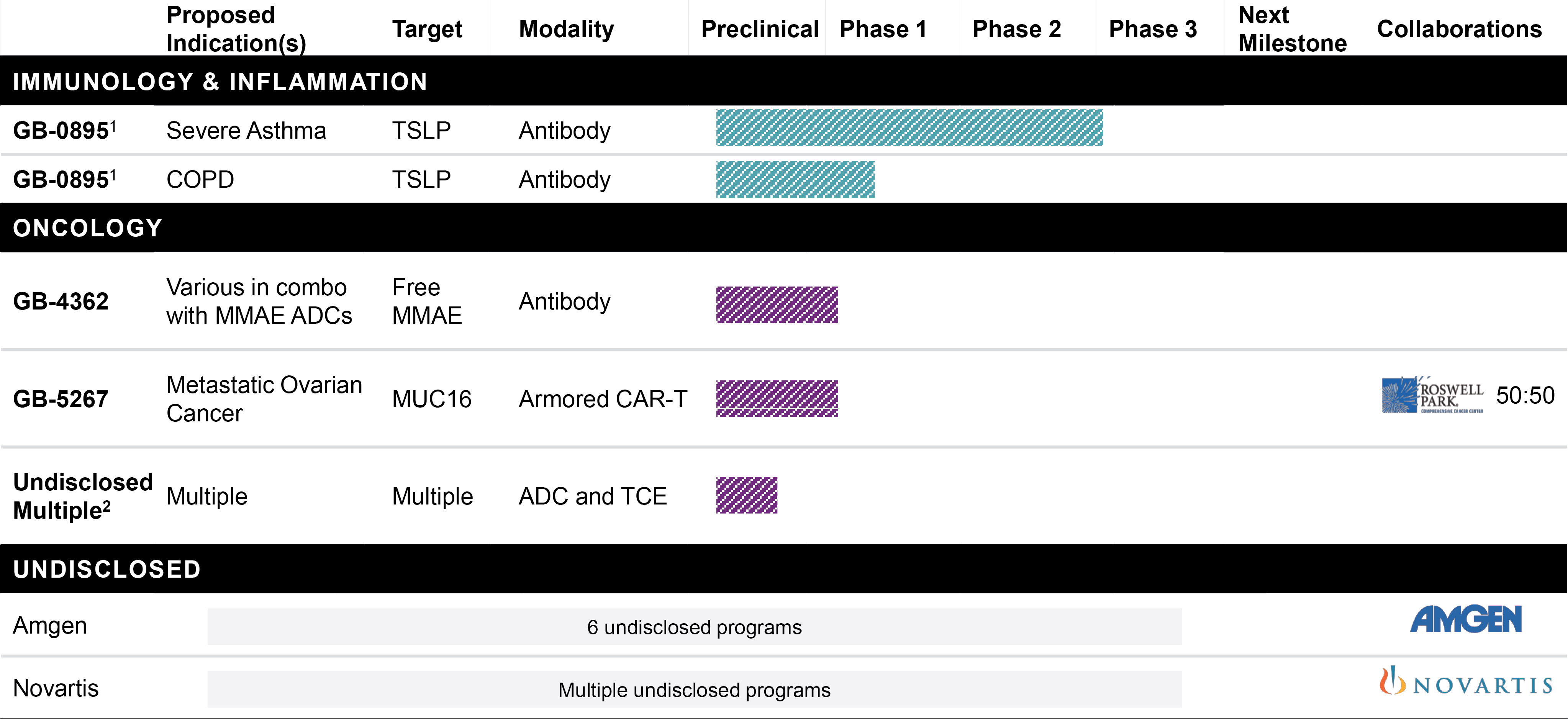


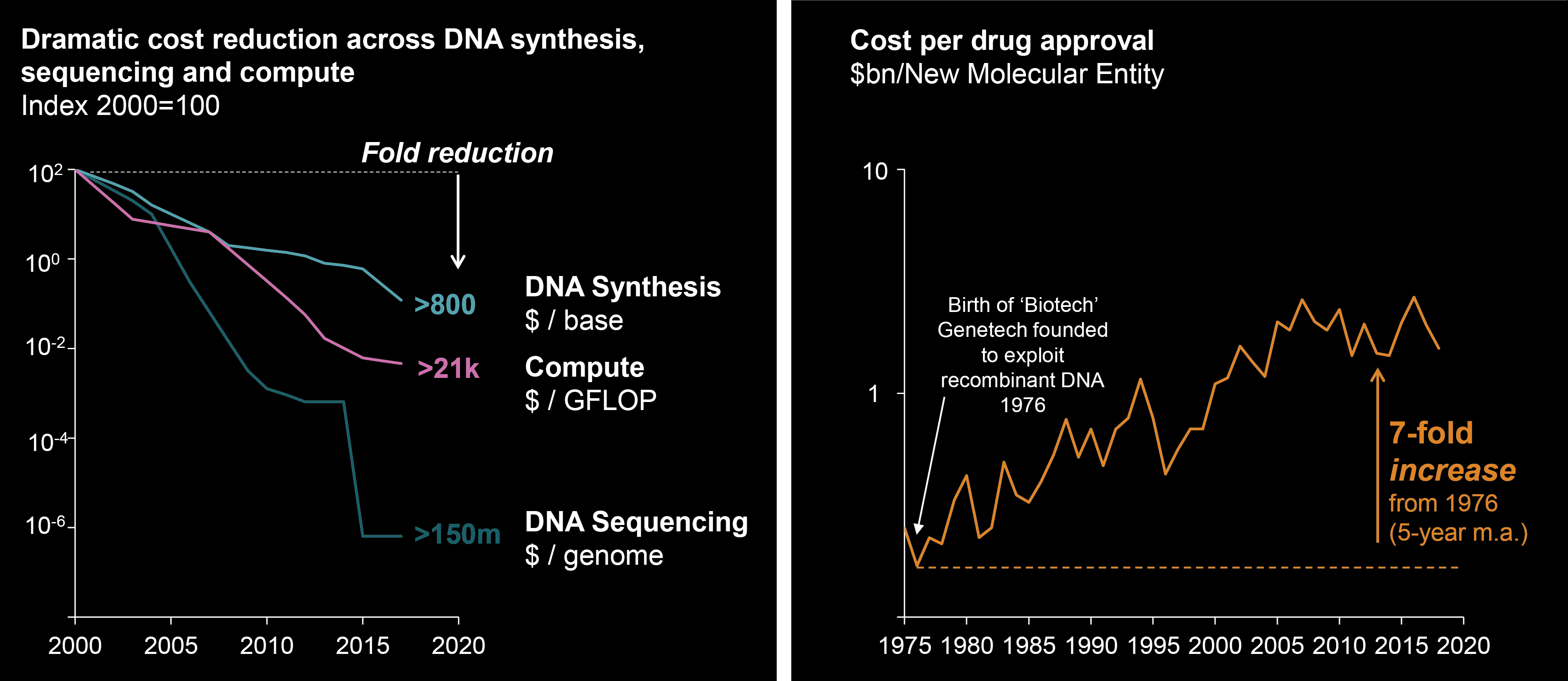

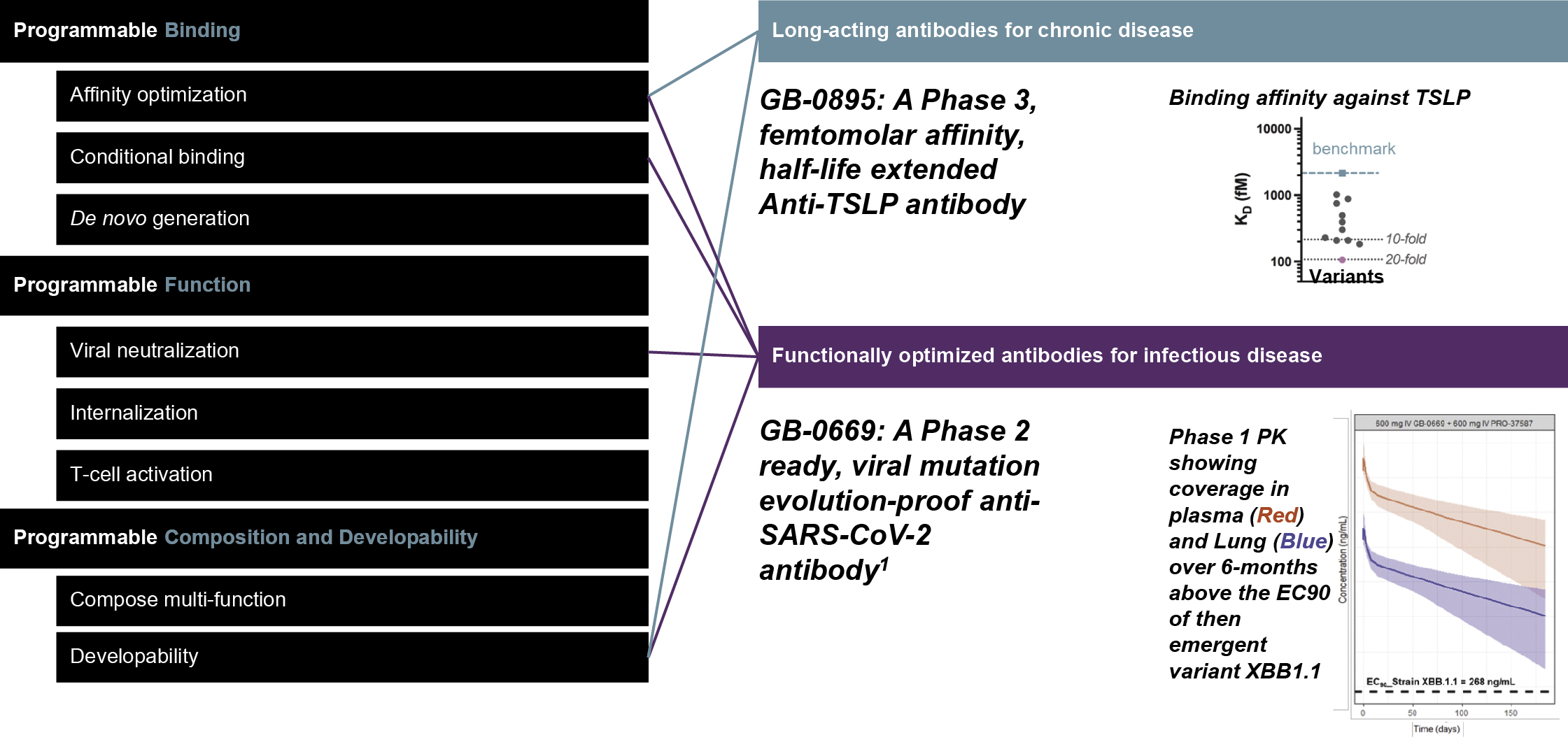
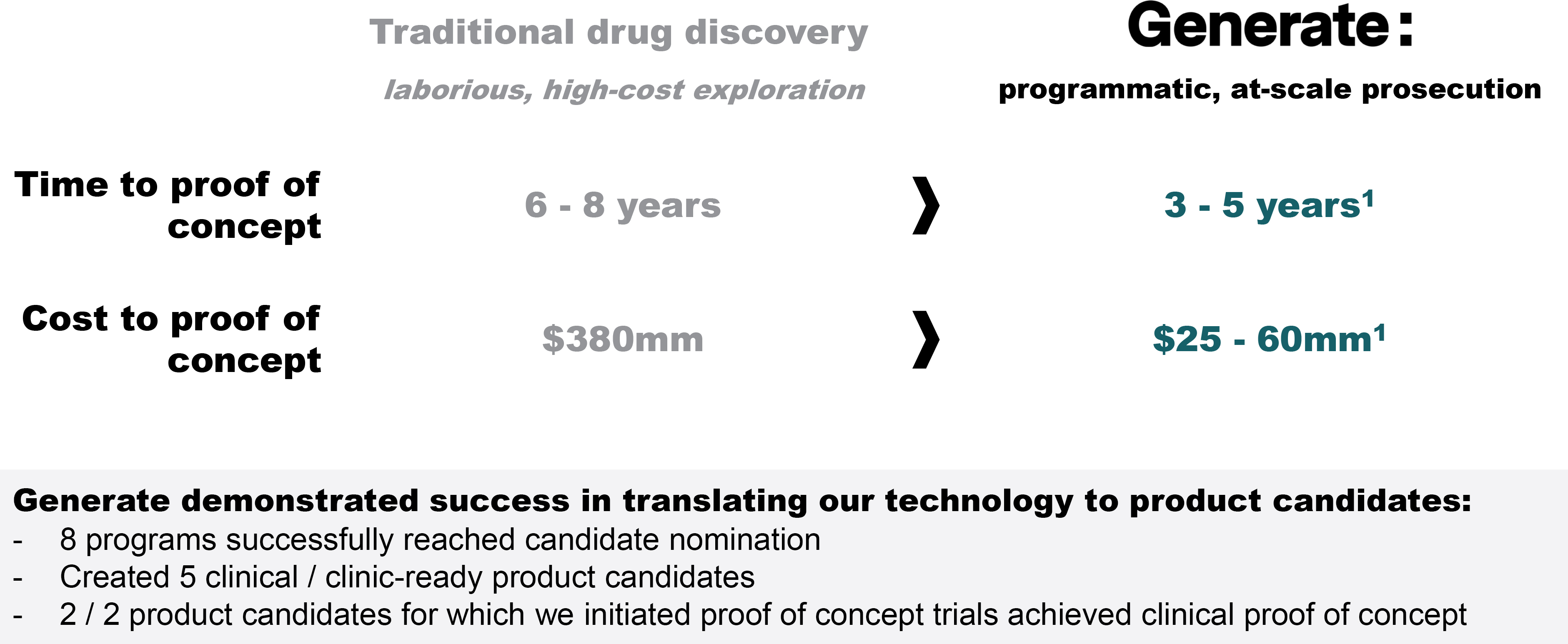
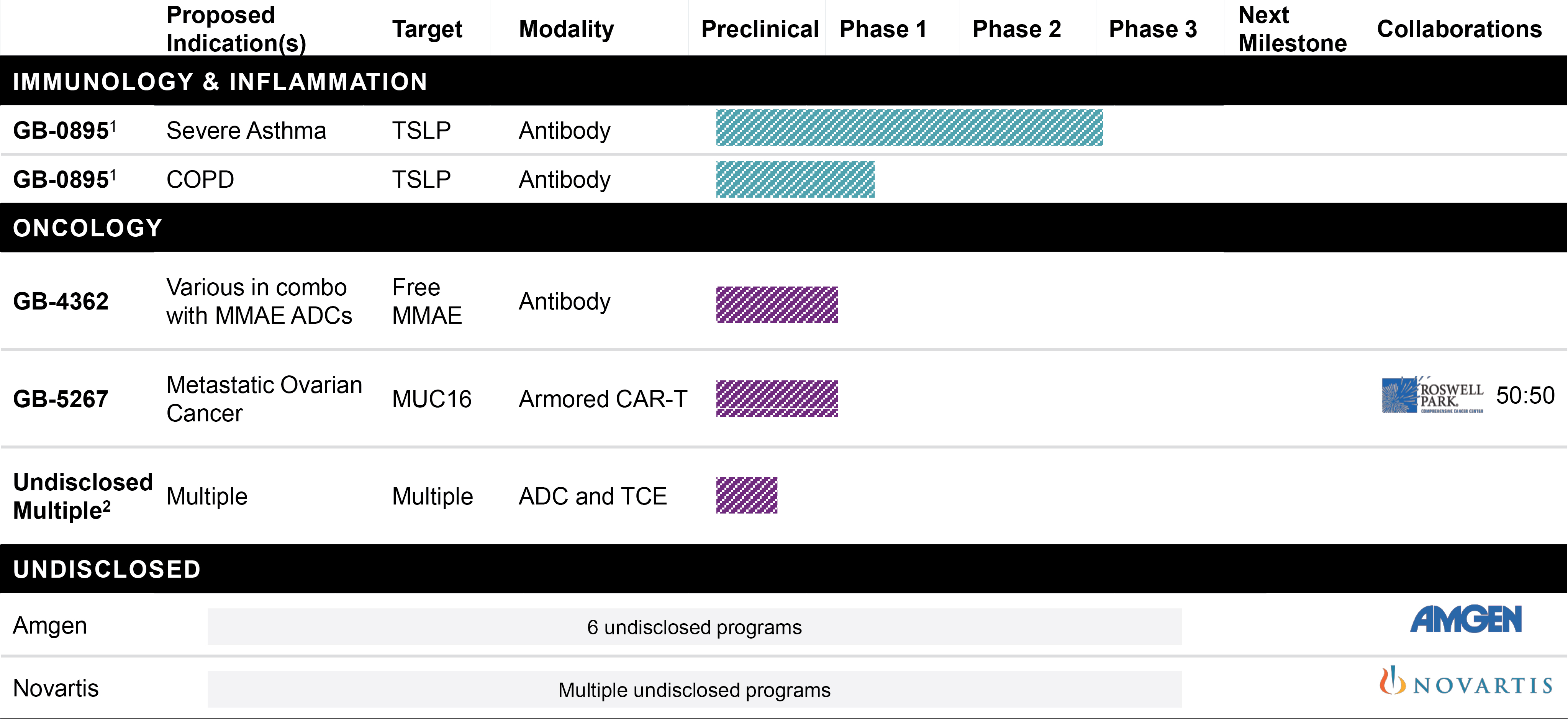
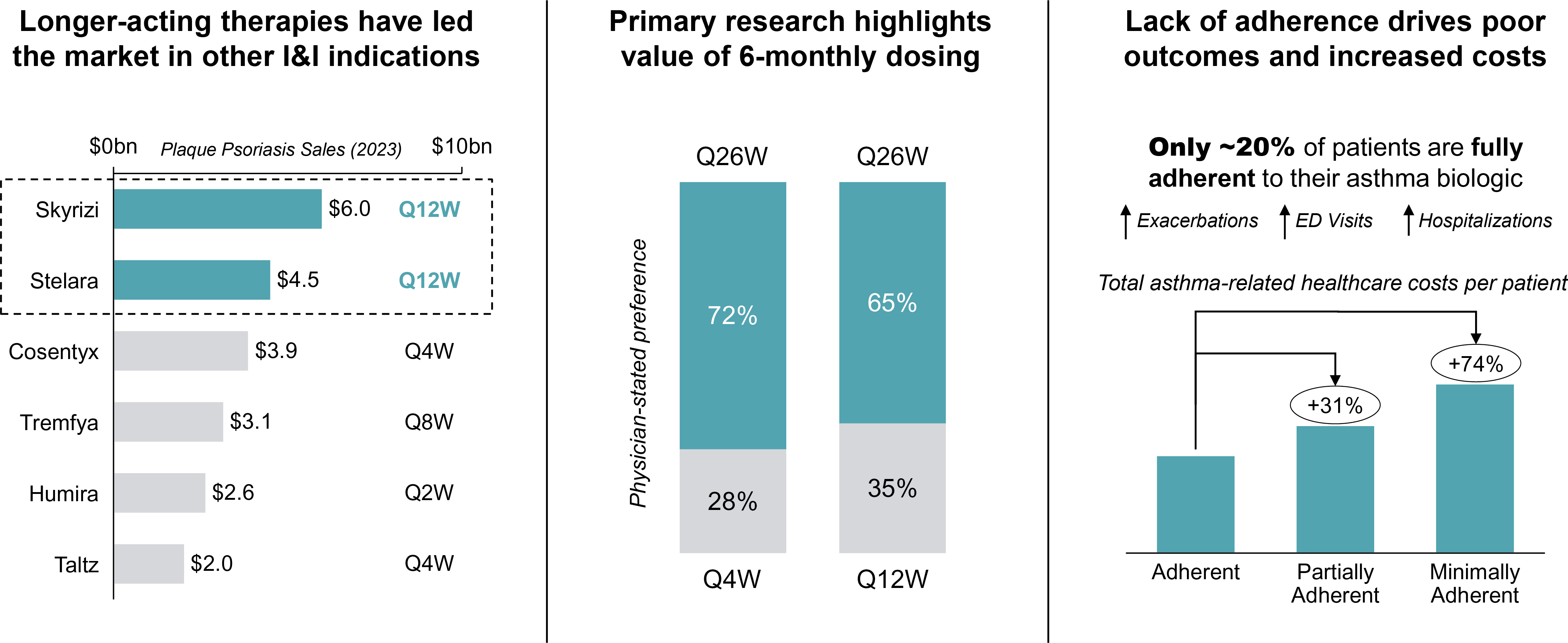
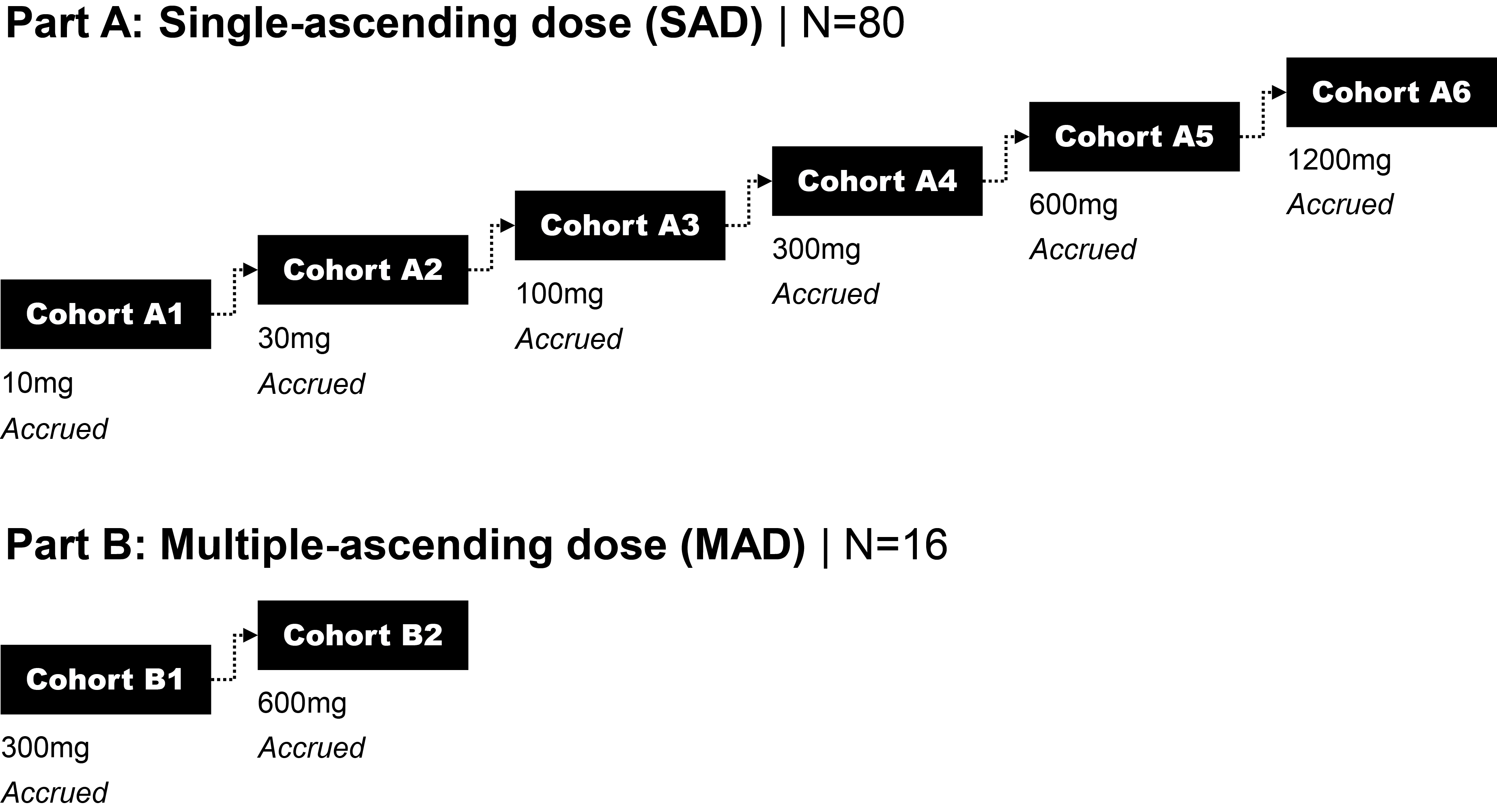
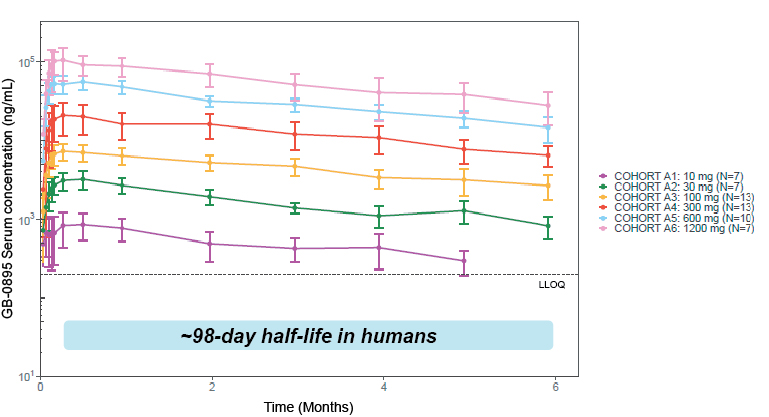
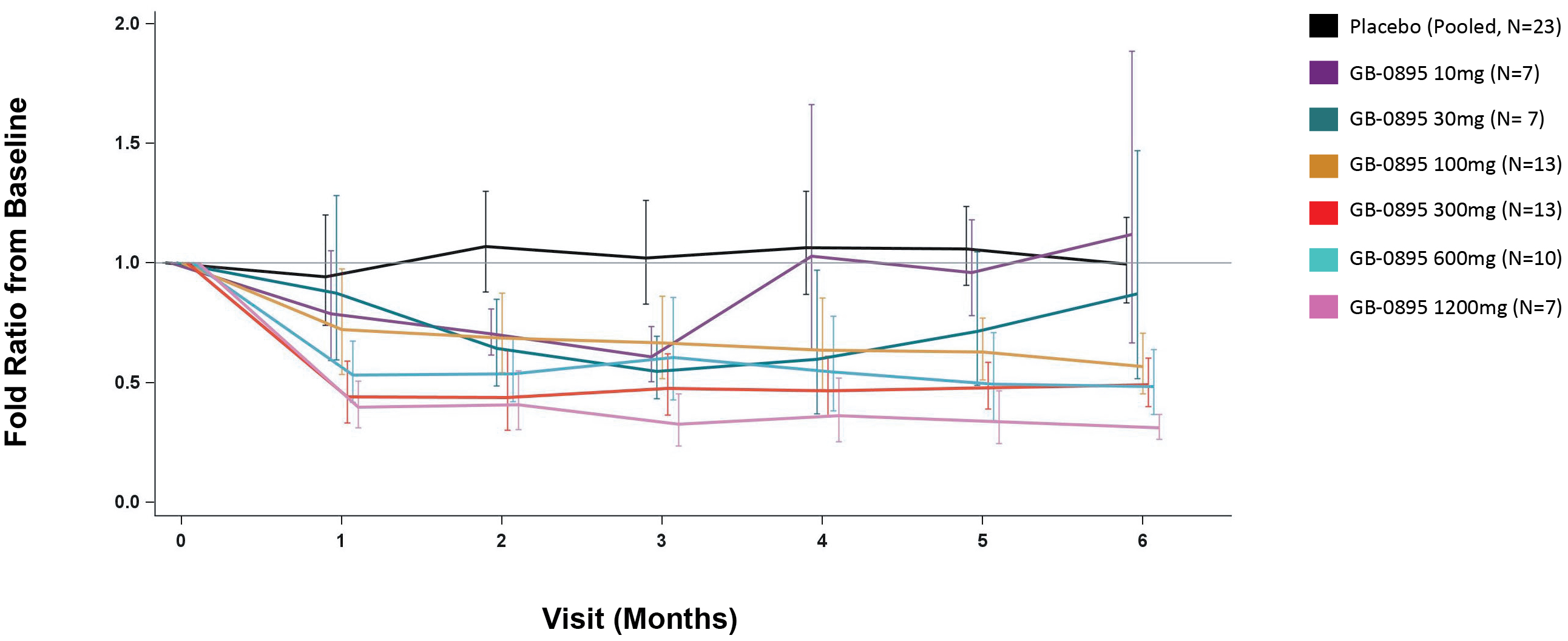
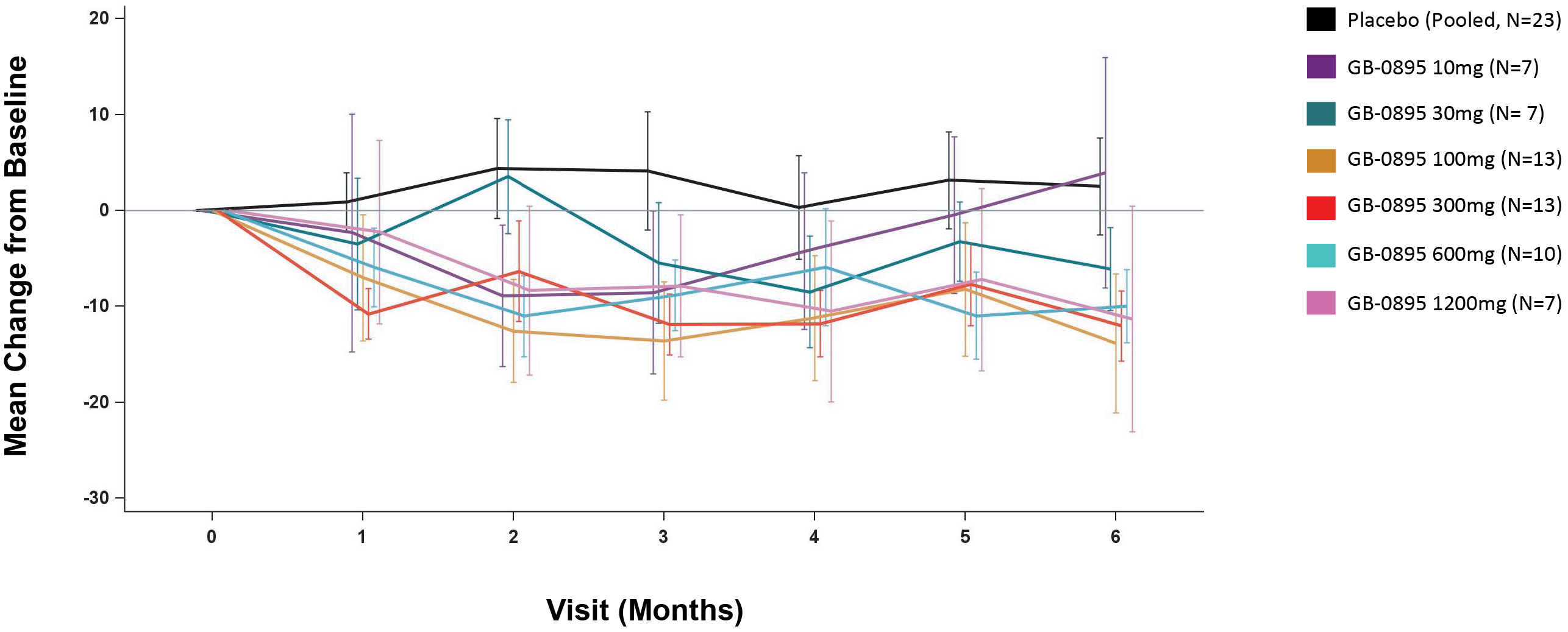
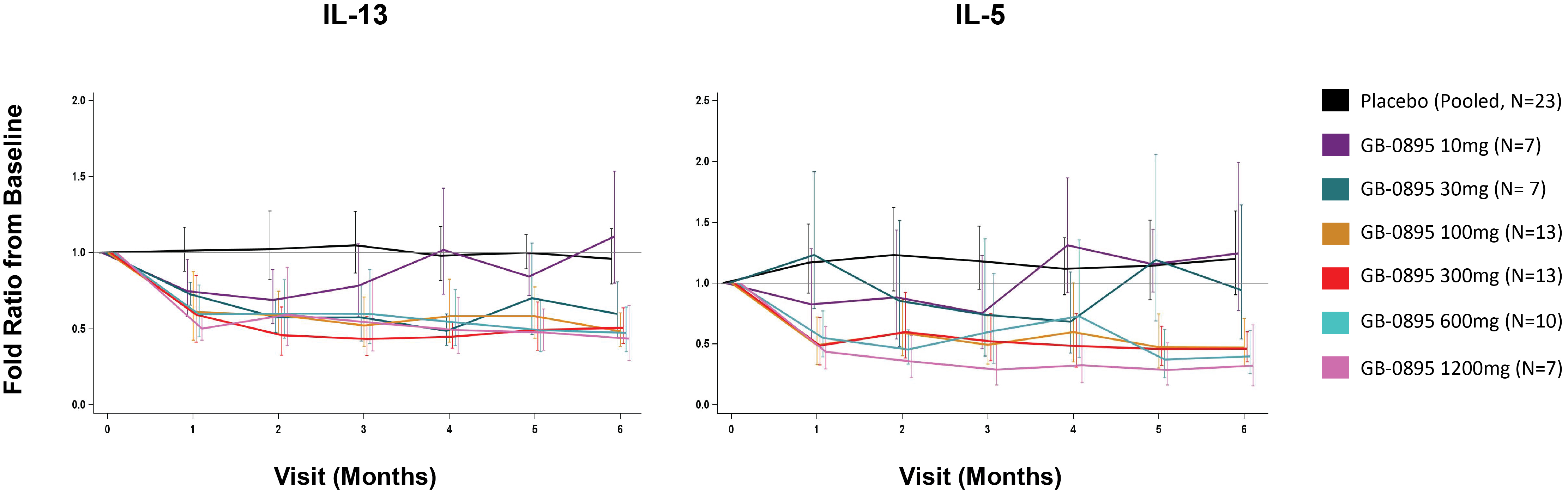
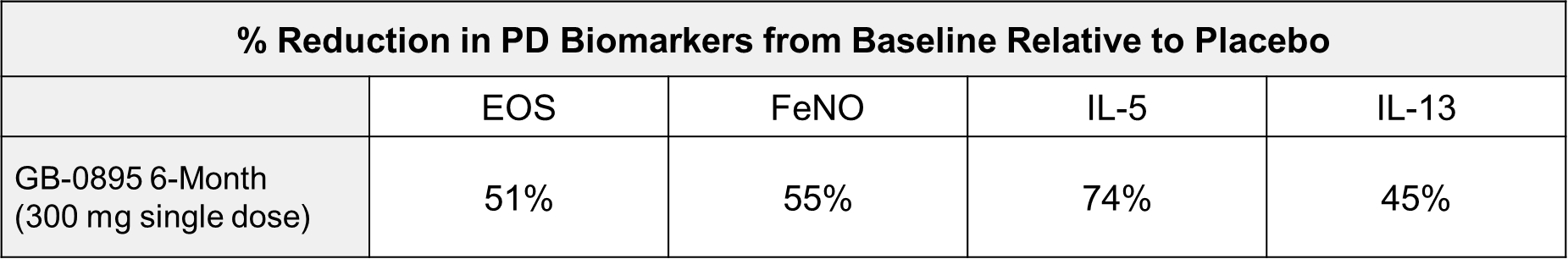
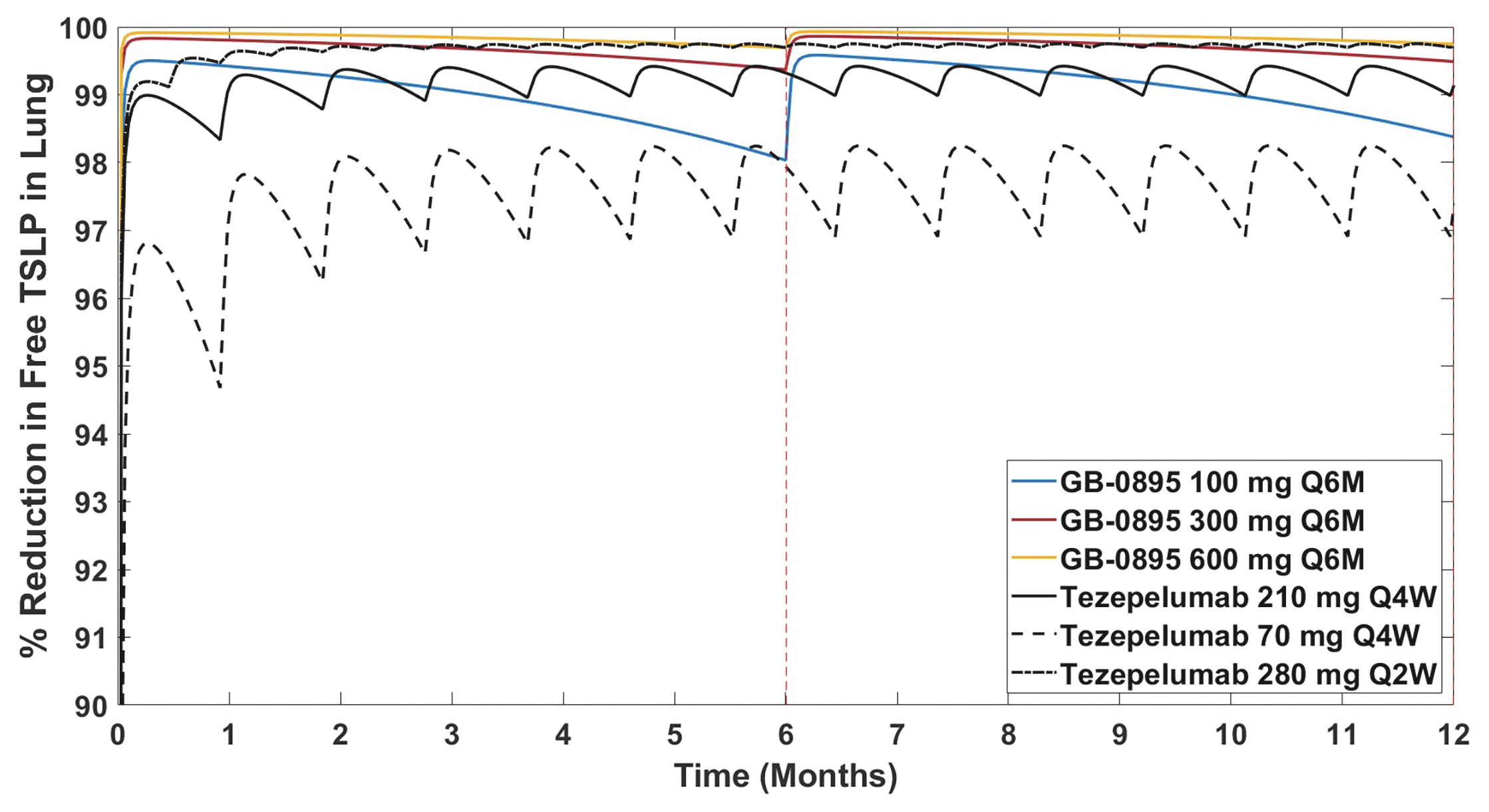
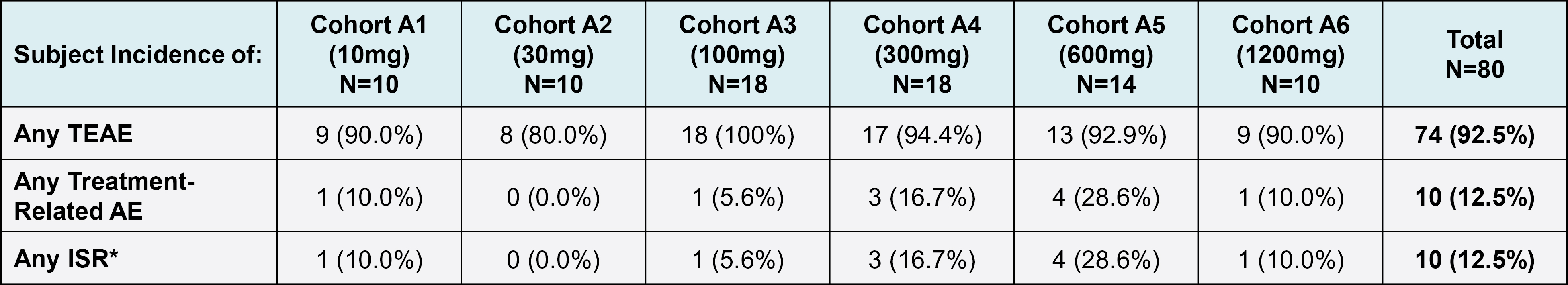
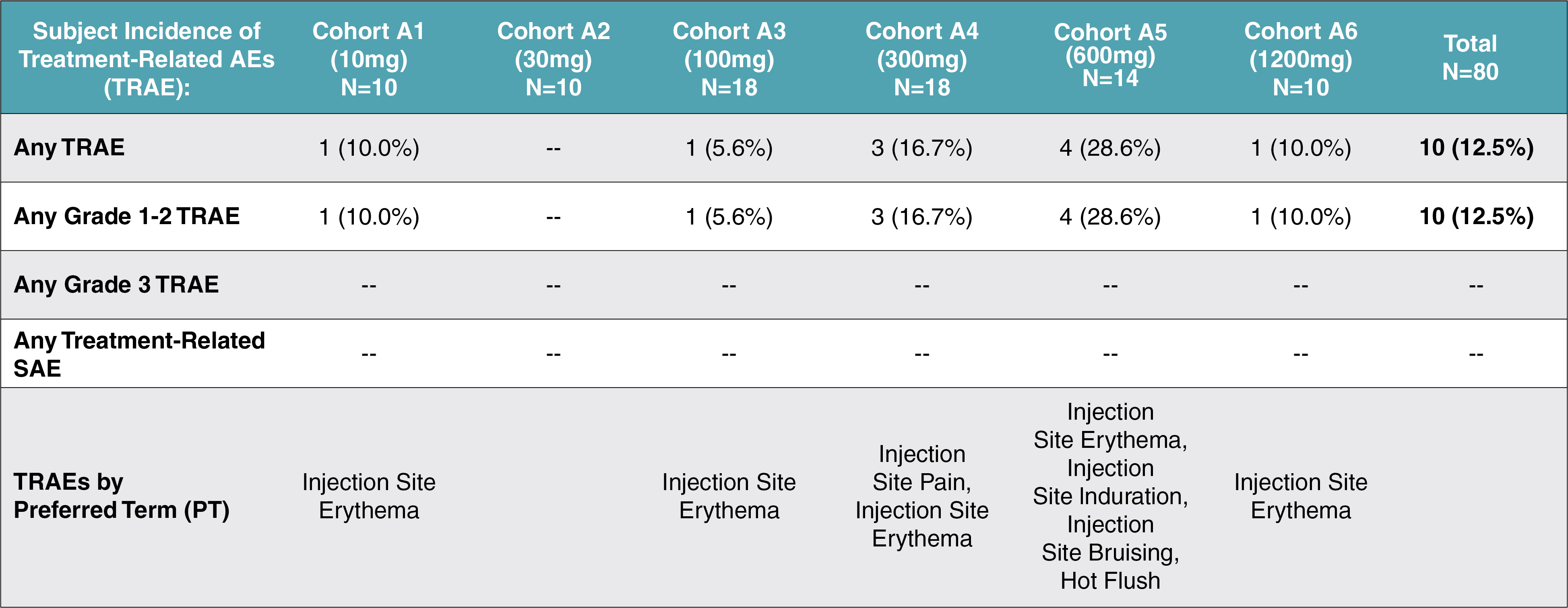
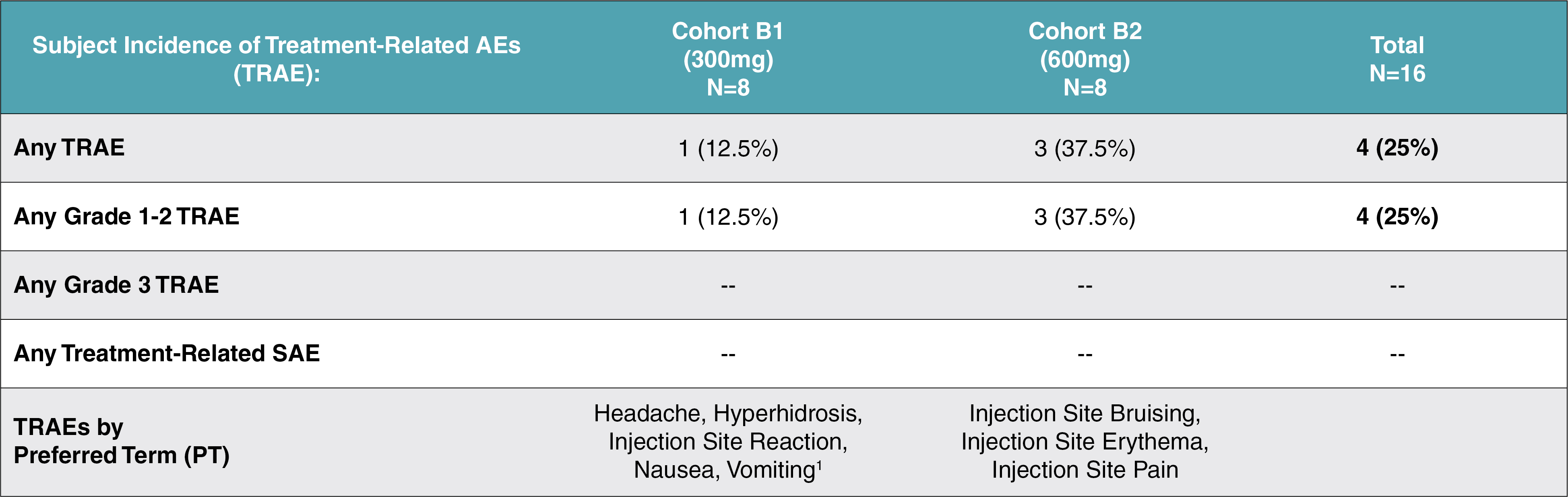
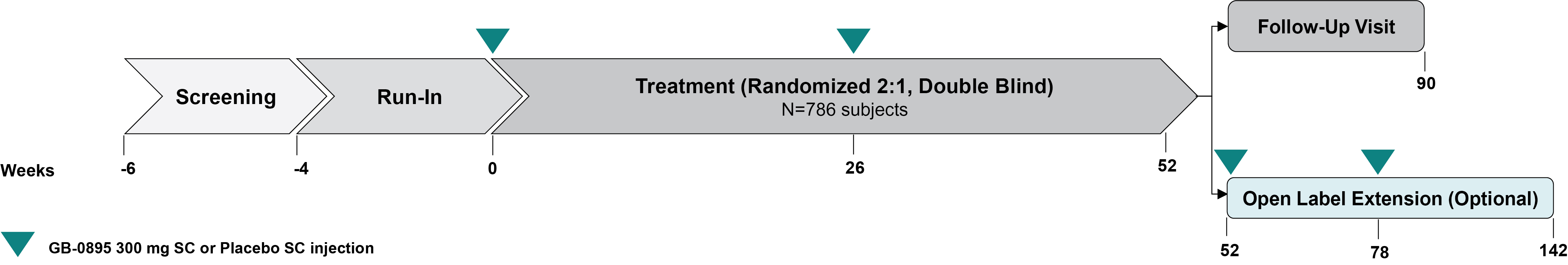
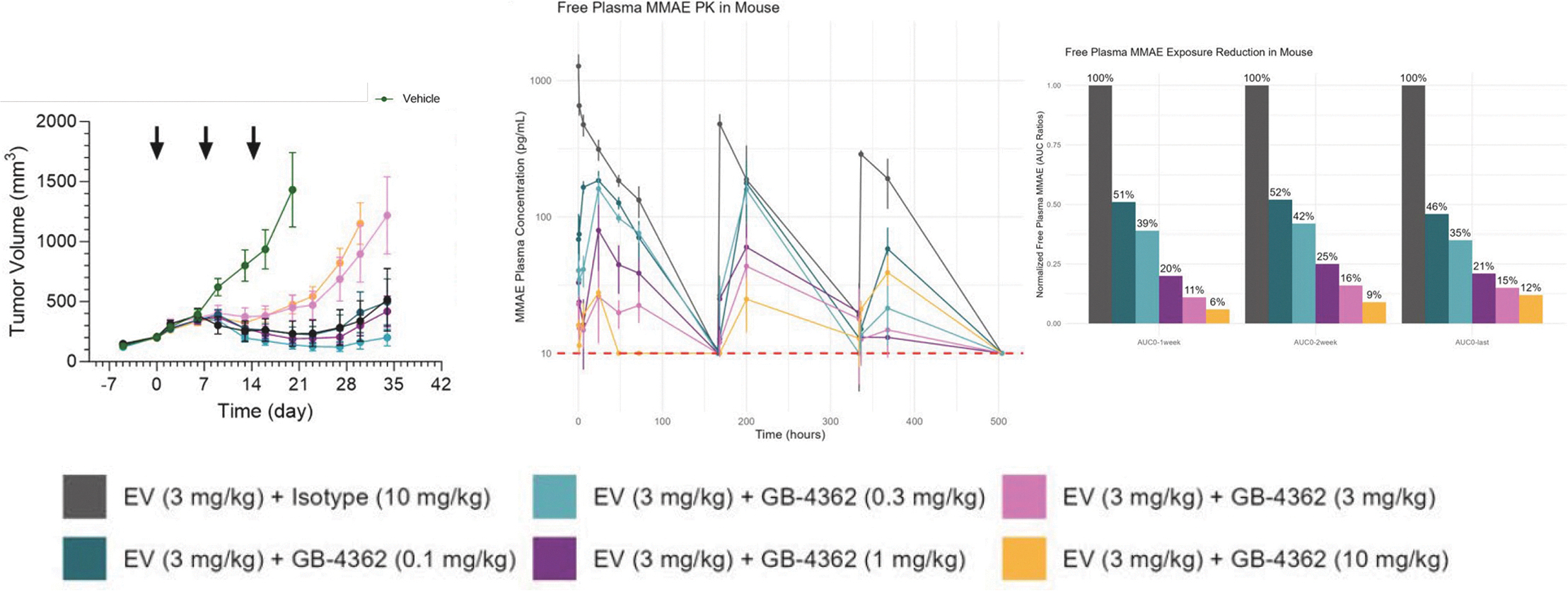
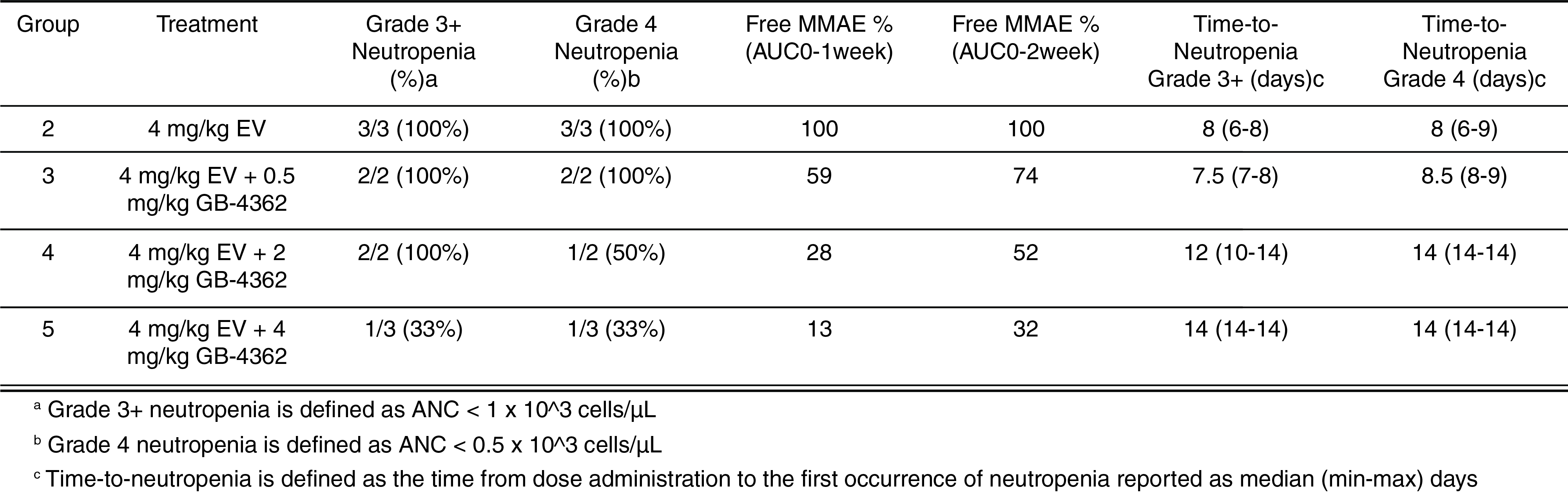
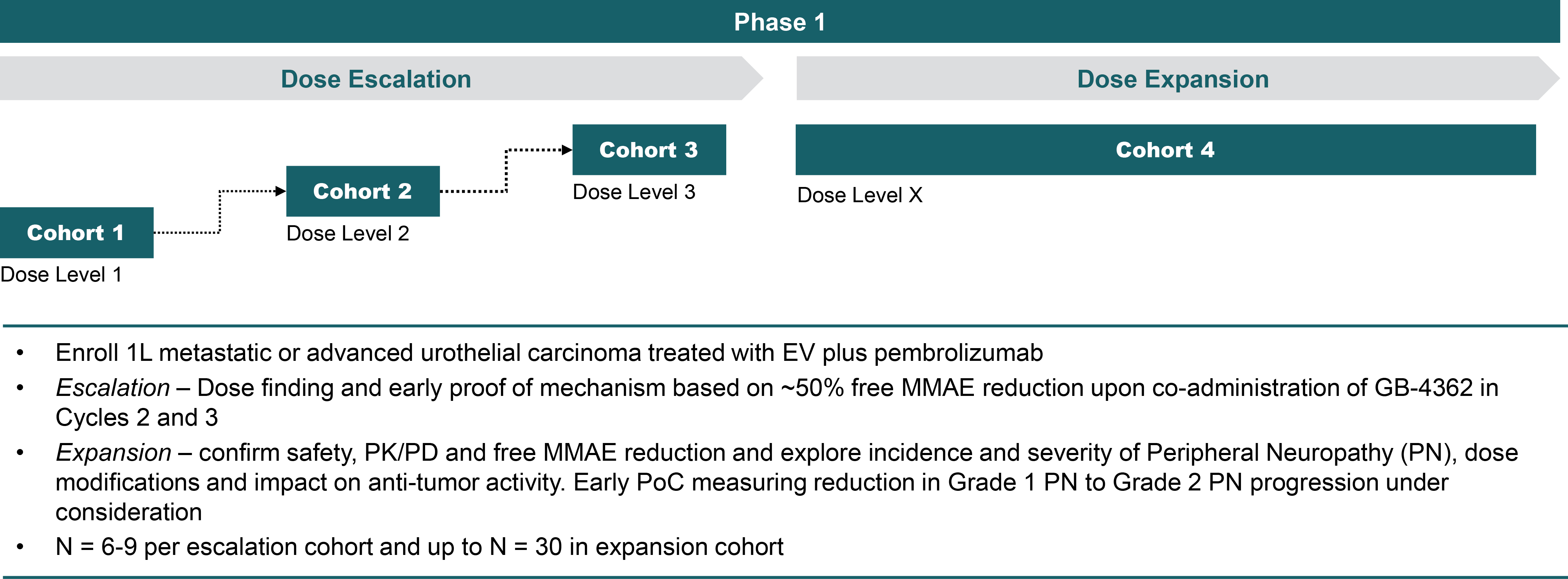
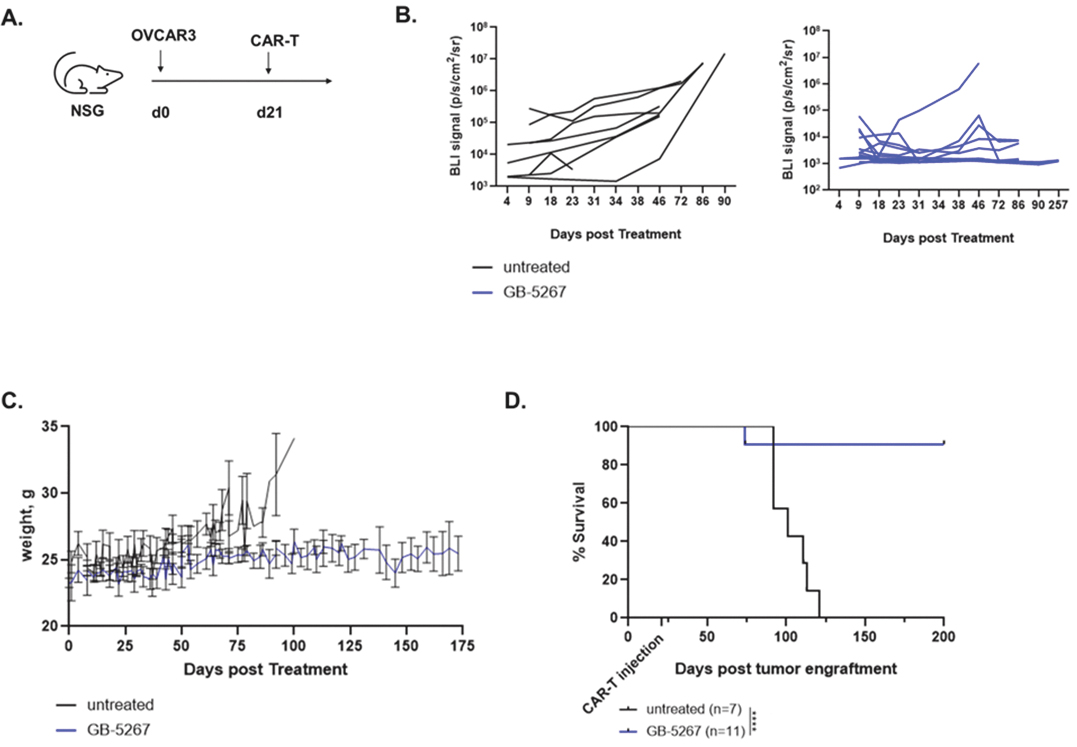
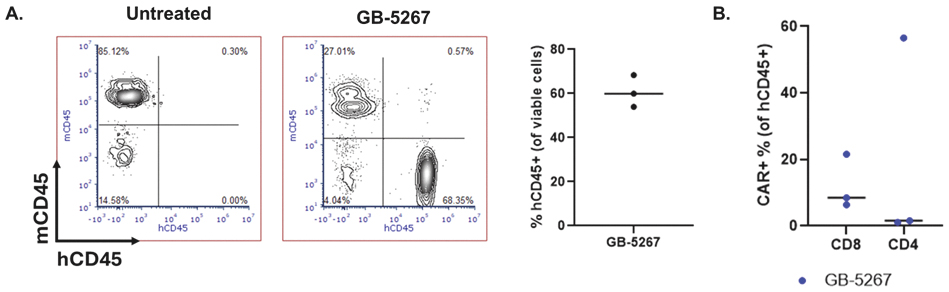
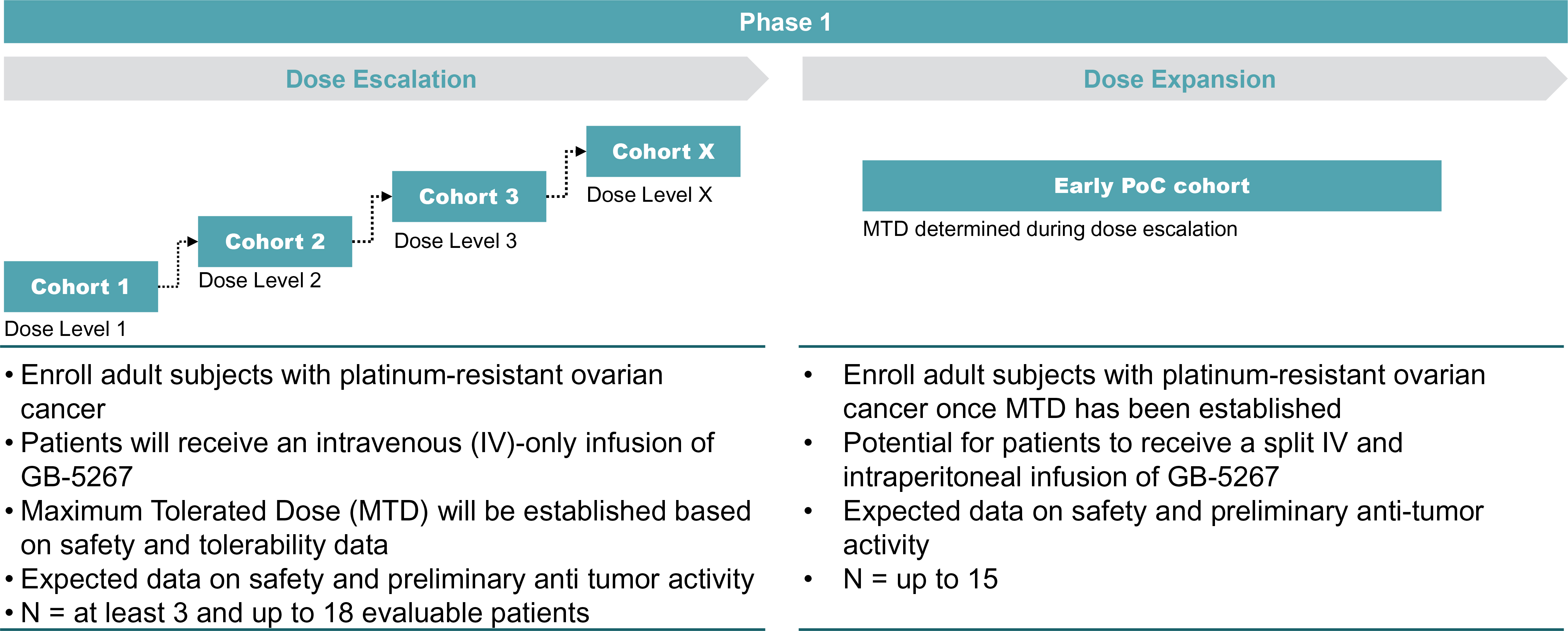
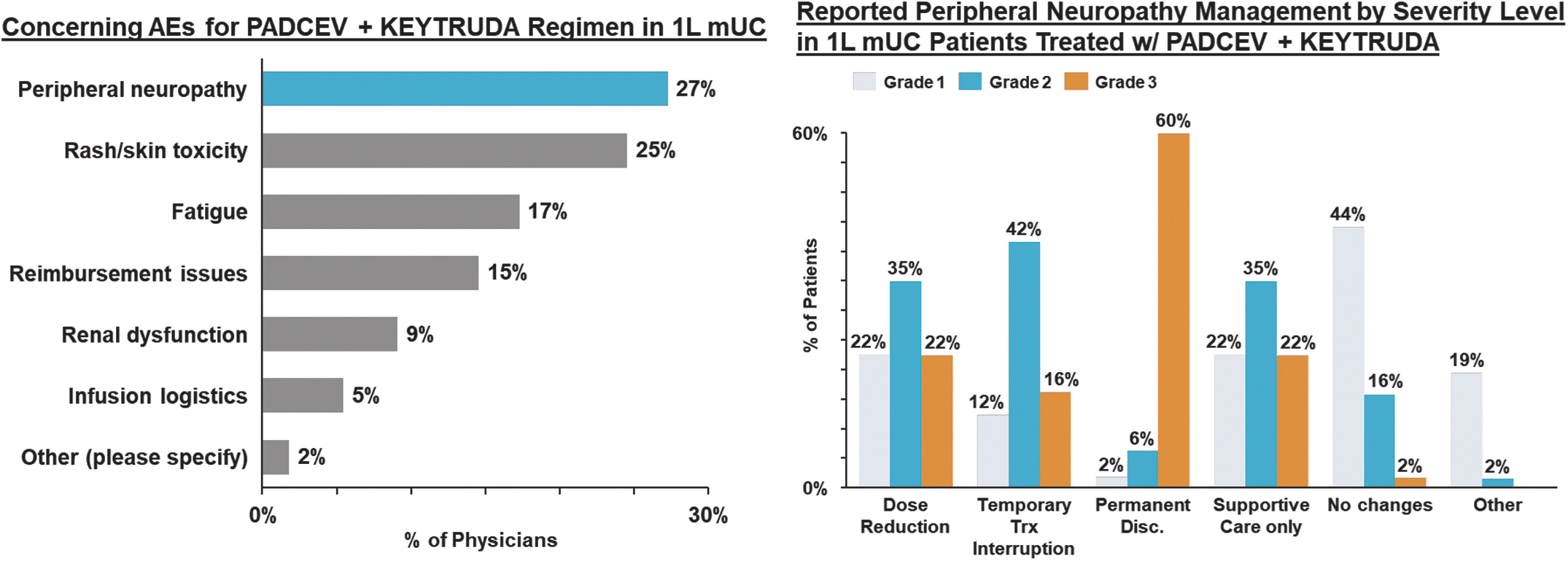 EV is marketed as PADCEV by Astellas and Pfizer Inc. ("Pfizer") and pembrolizumab is marketed as KEYTRUDA by Merck.
EV is marketed as PADCEV by Astellas and Pfizer Inc. ("Pfizer") and pembrolizumab is marketed as KEYTRUDA by Merck.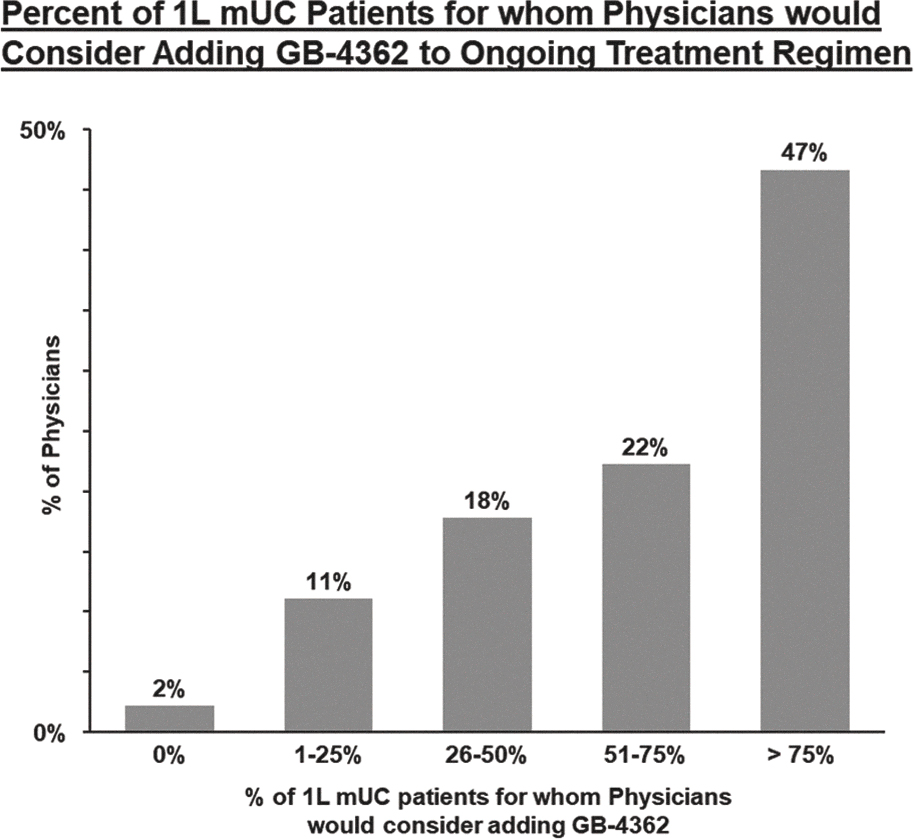
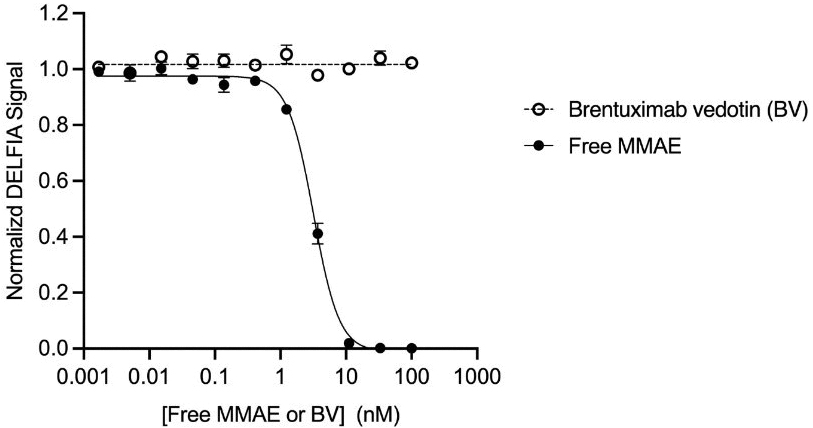
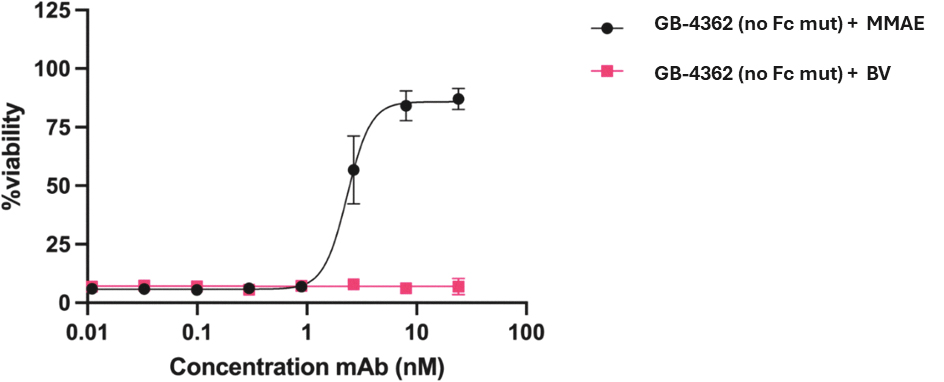
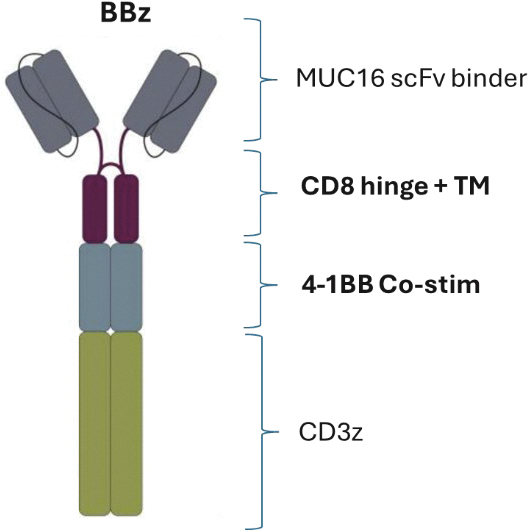
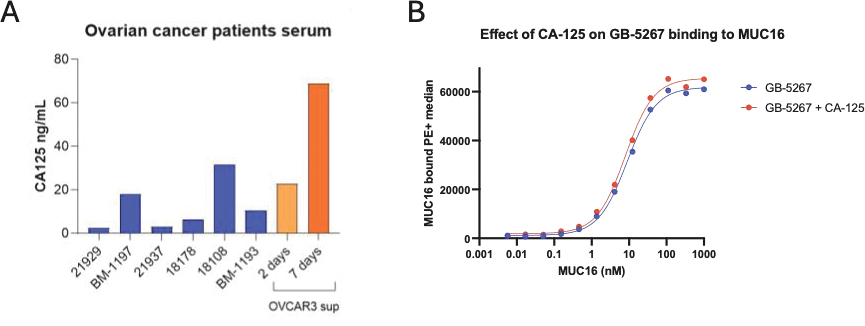
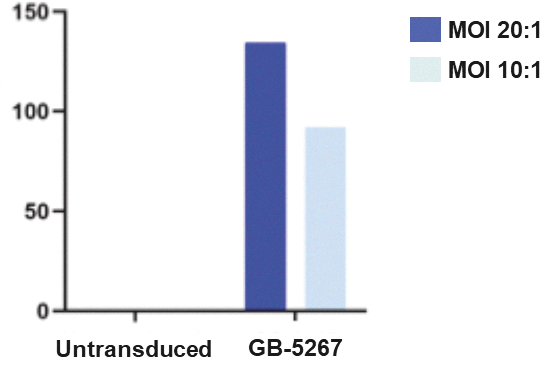
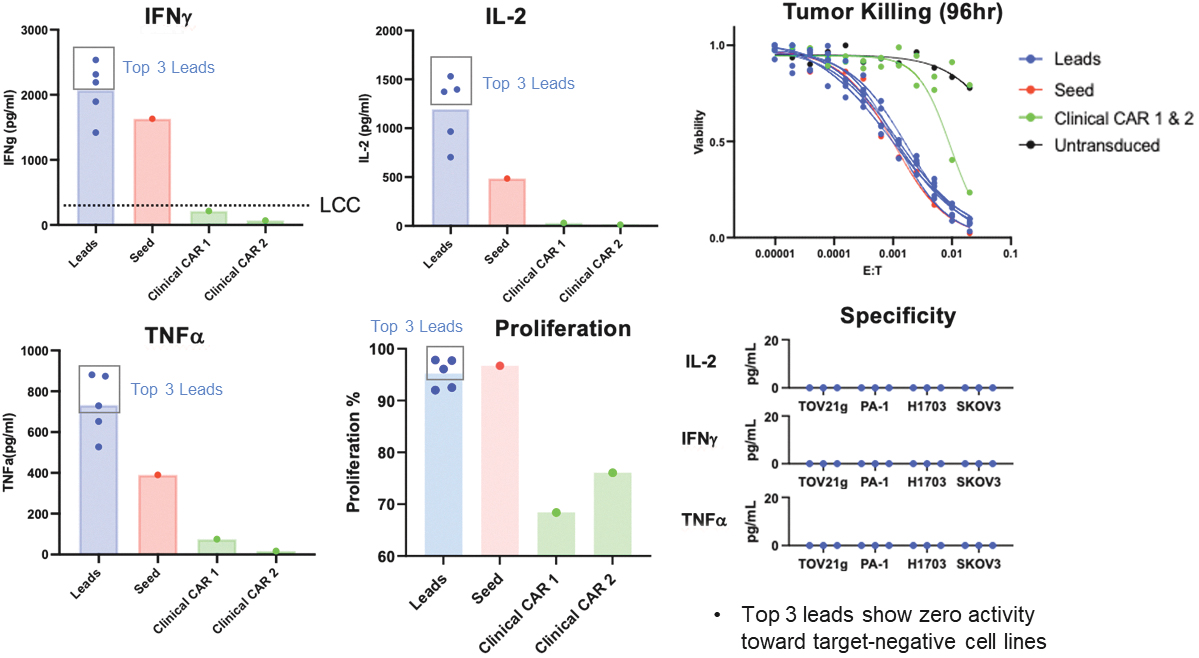
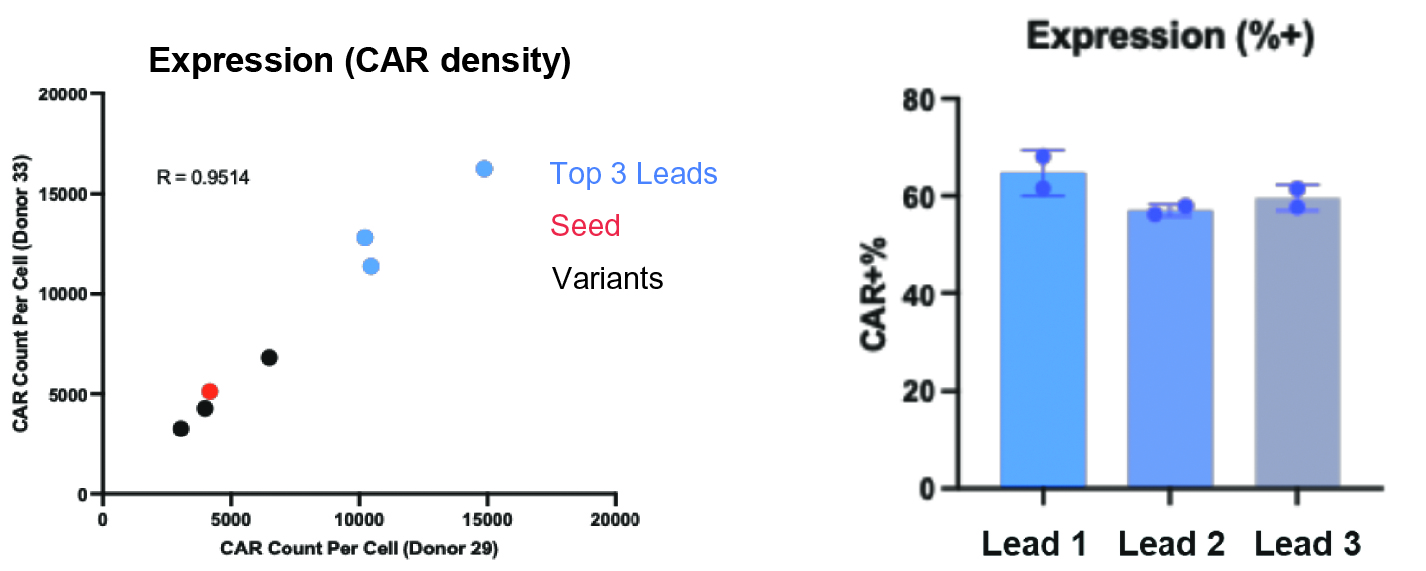
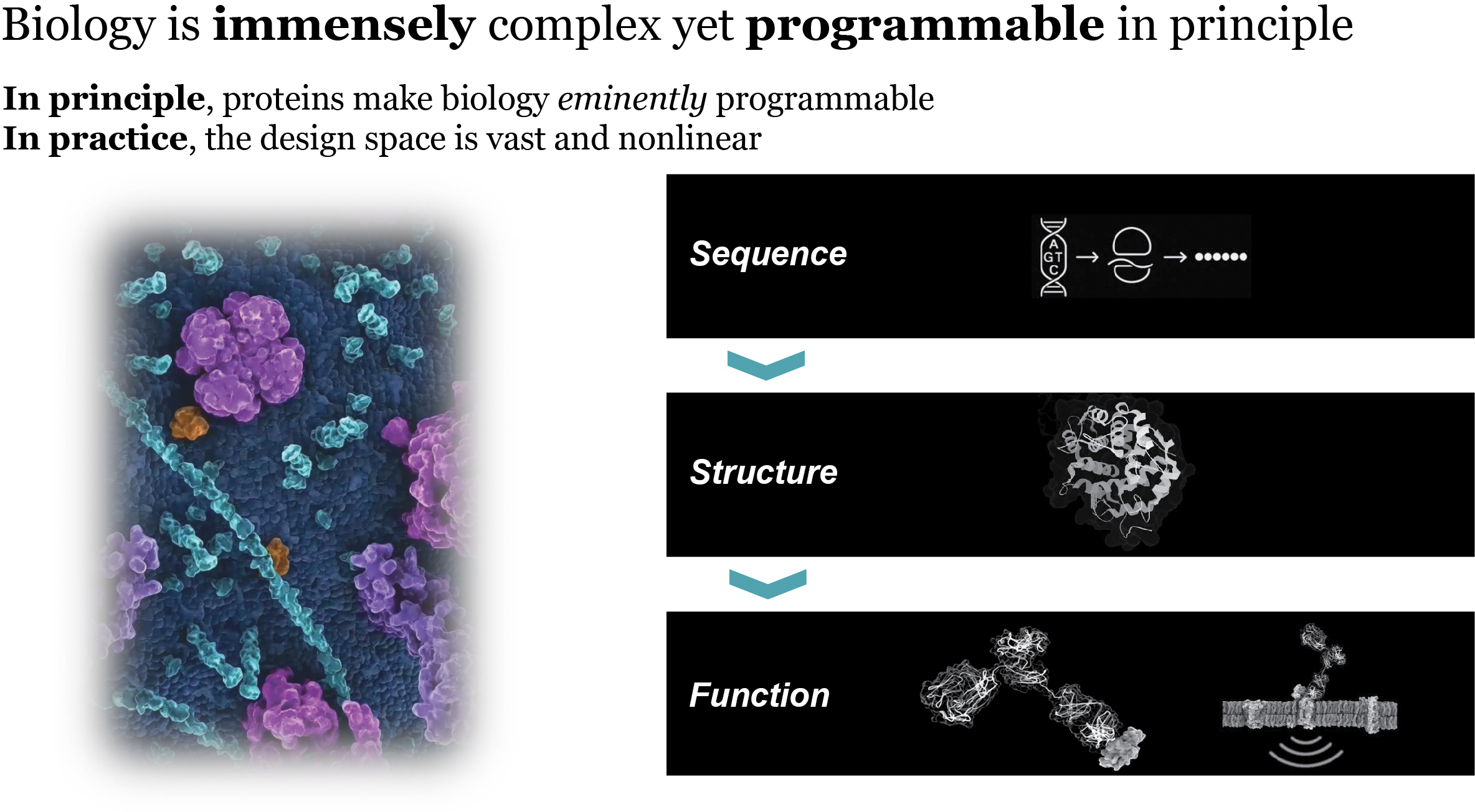
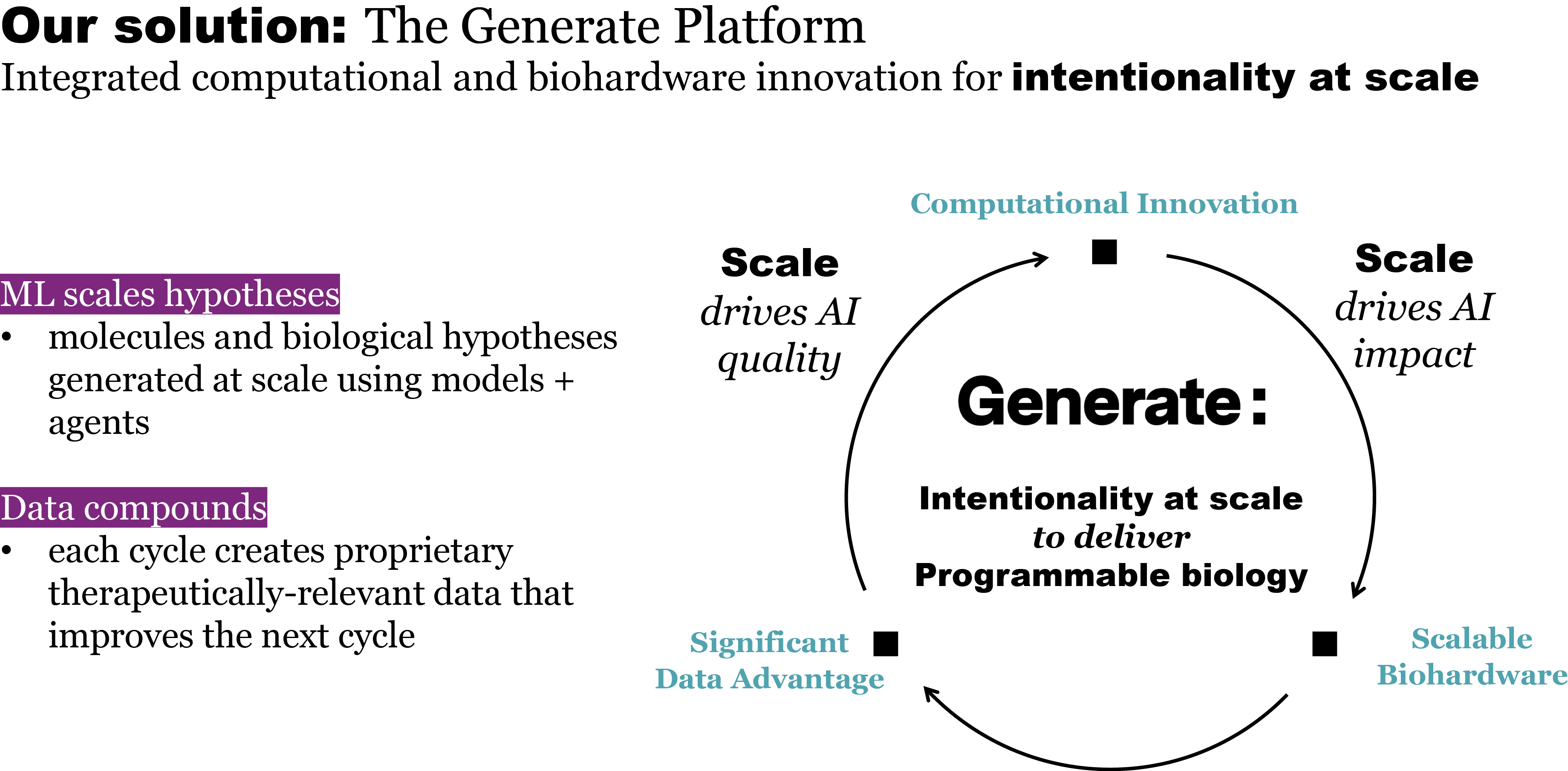

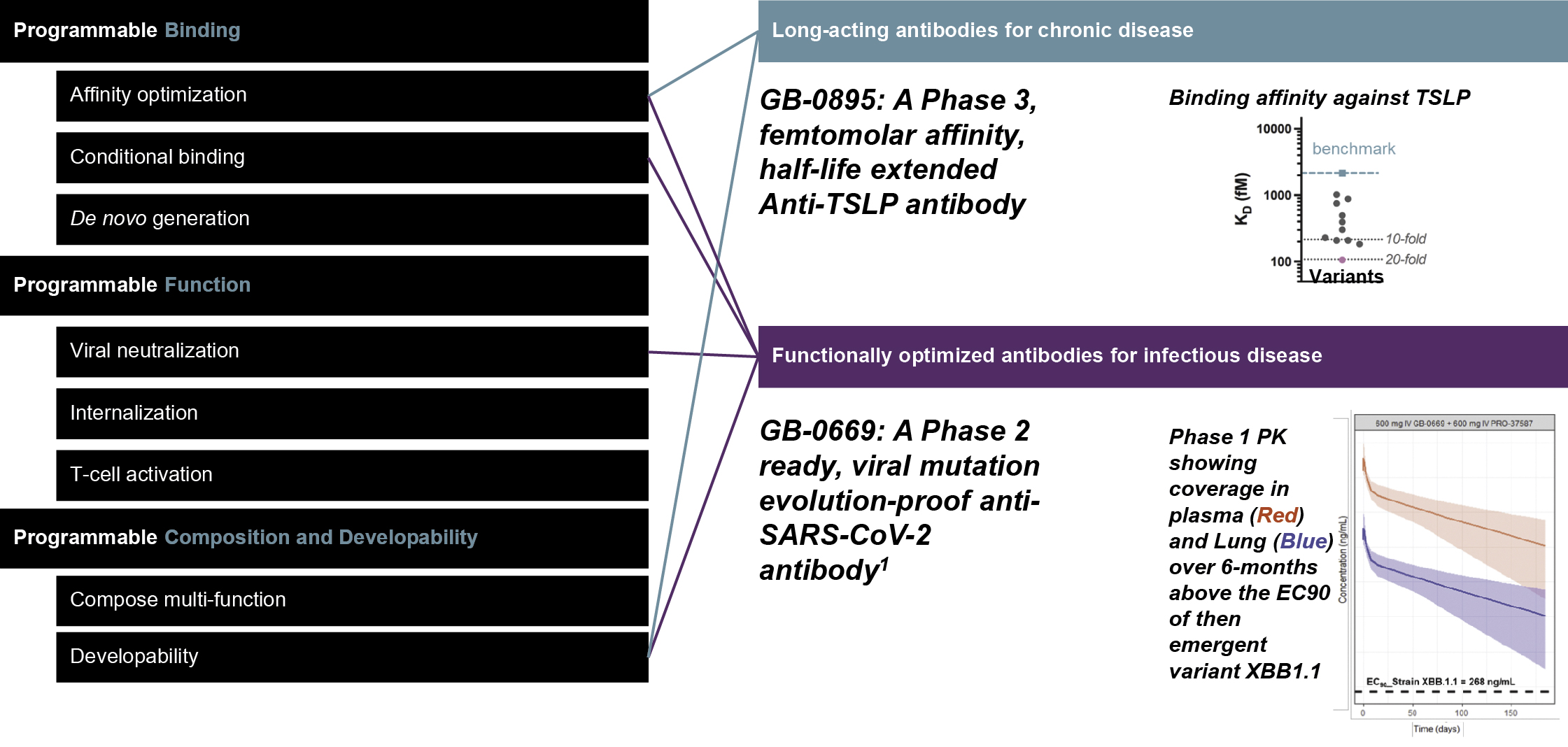
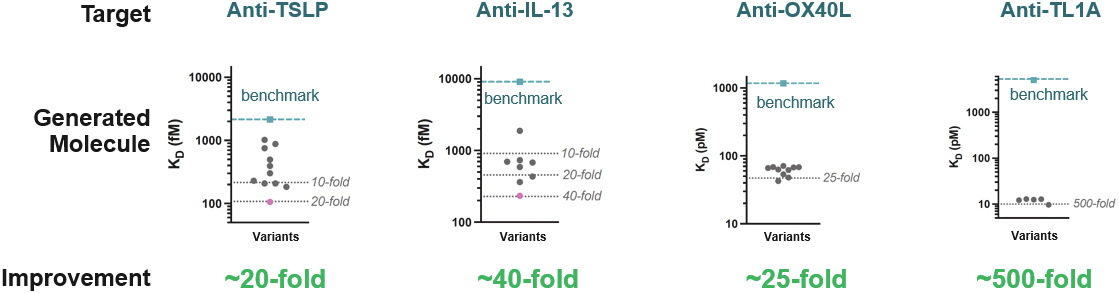
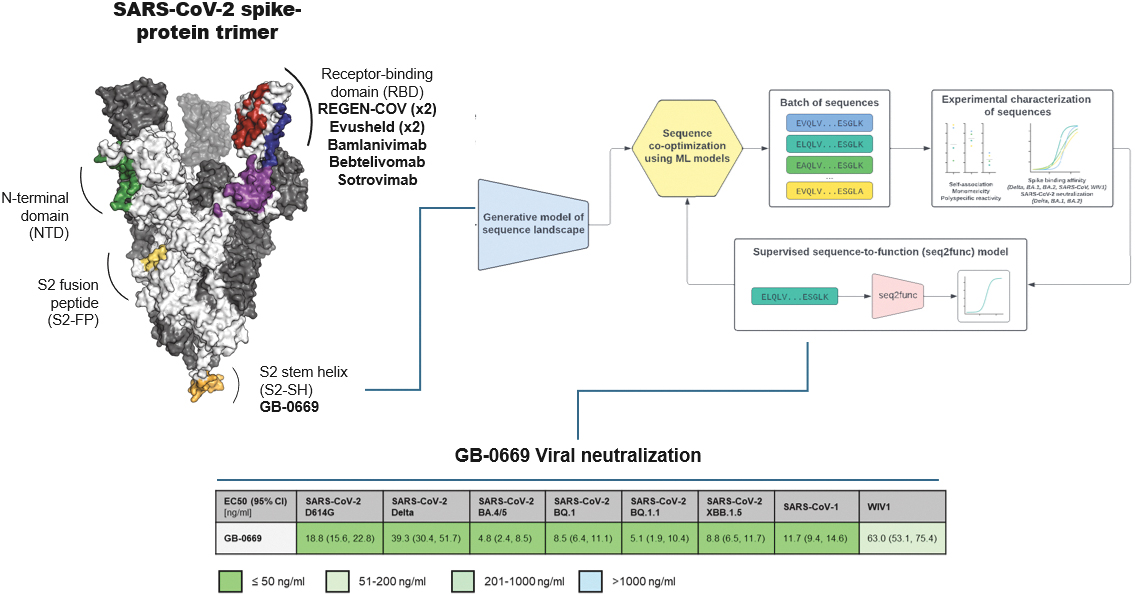

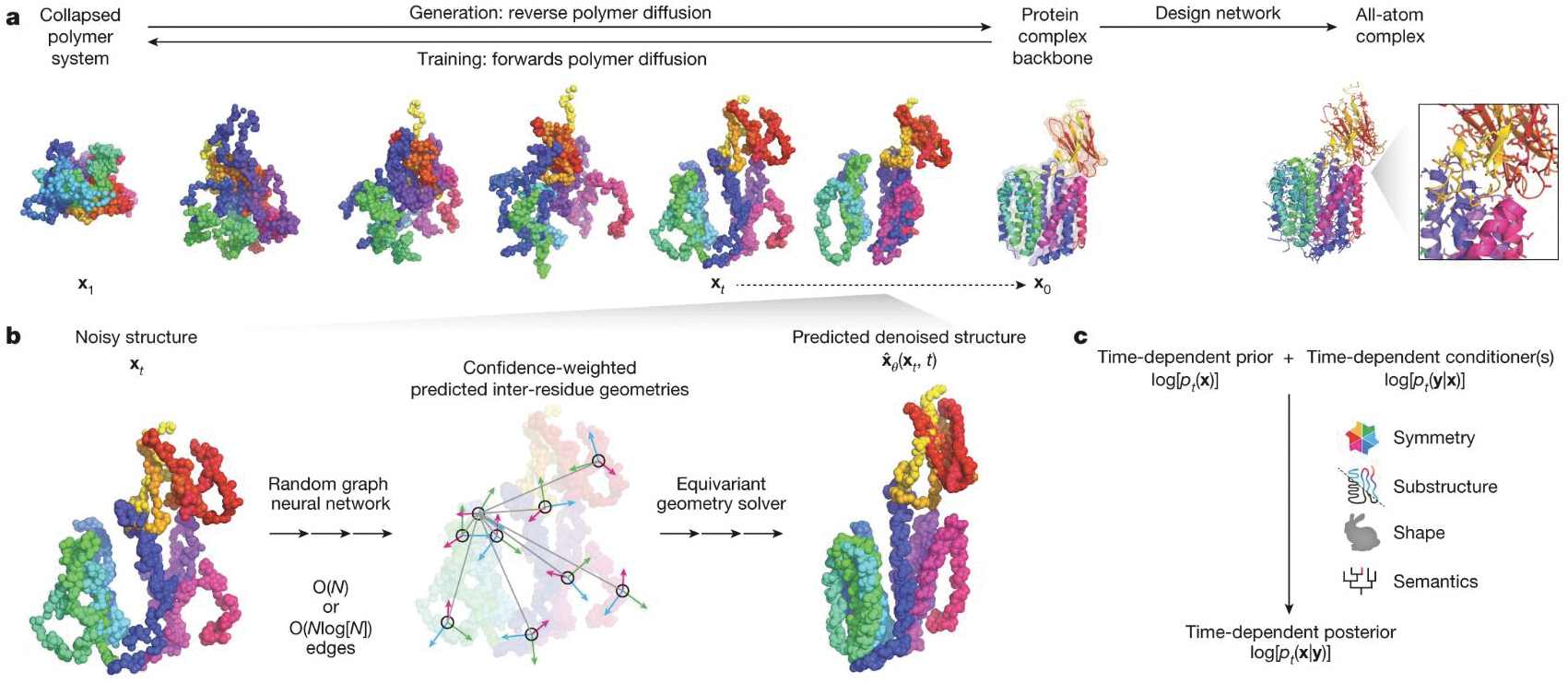
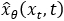 that maps noisy coordinates
that maps noisy coordinates 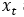 at time t to predicted denoised coordinates
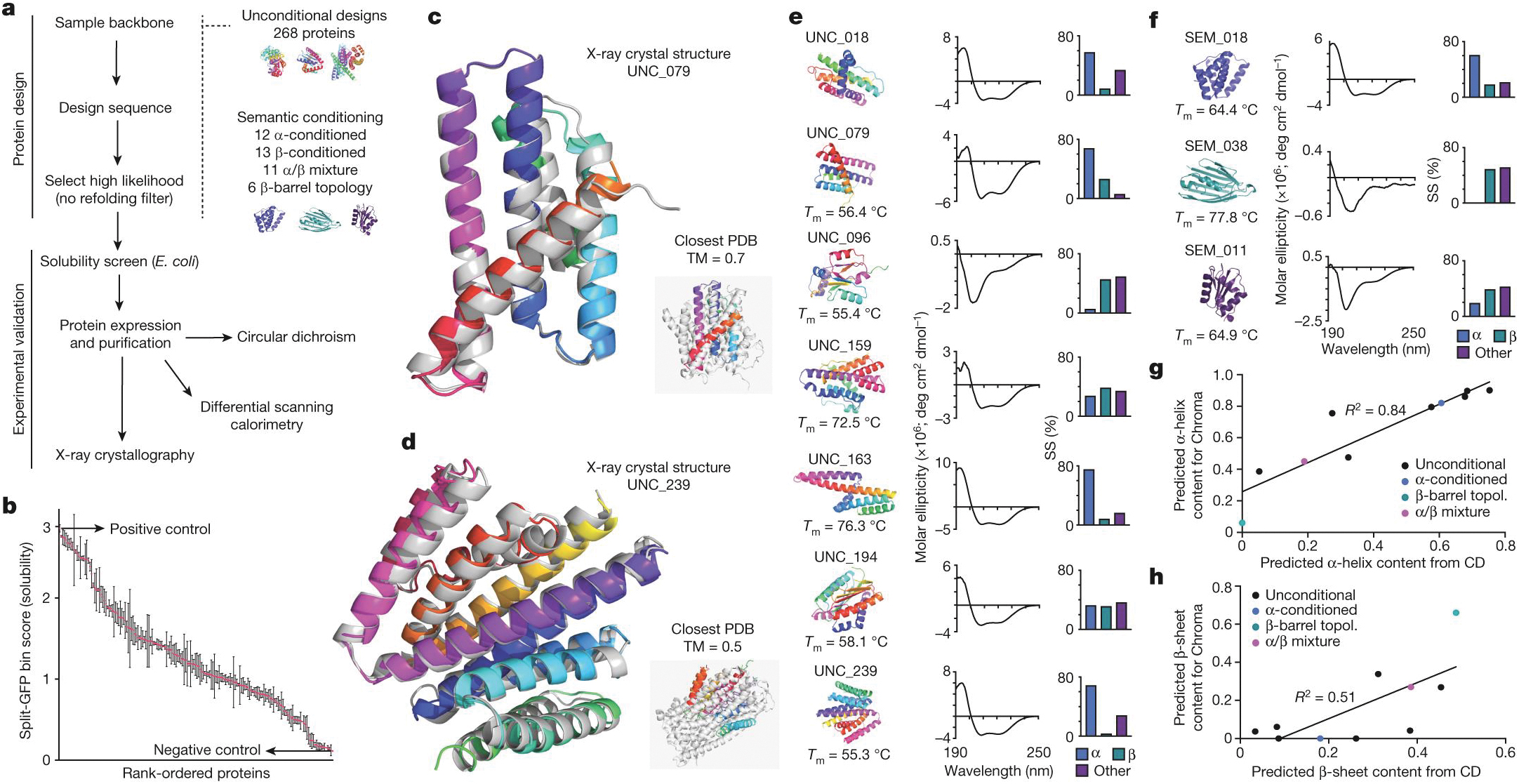
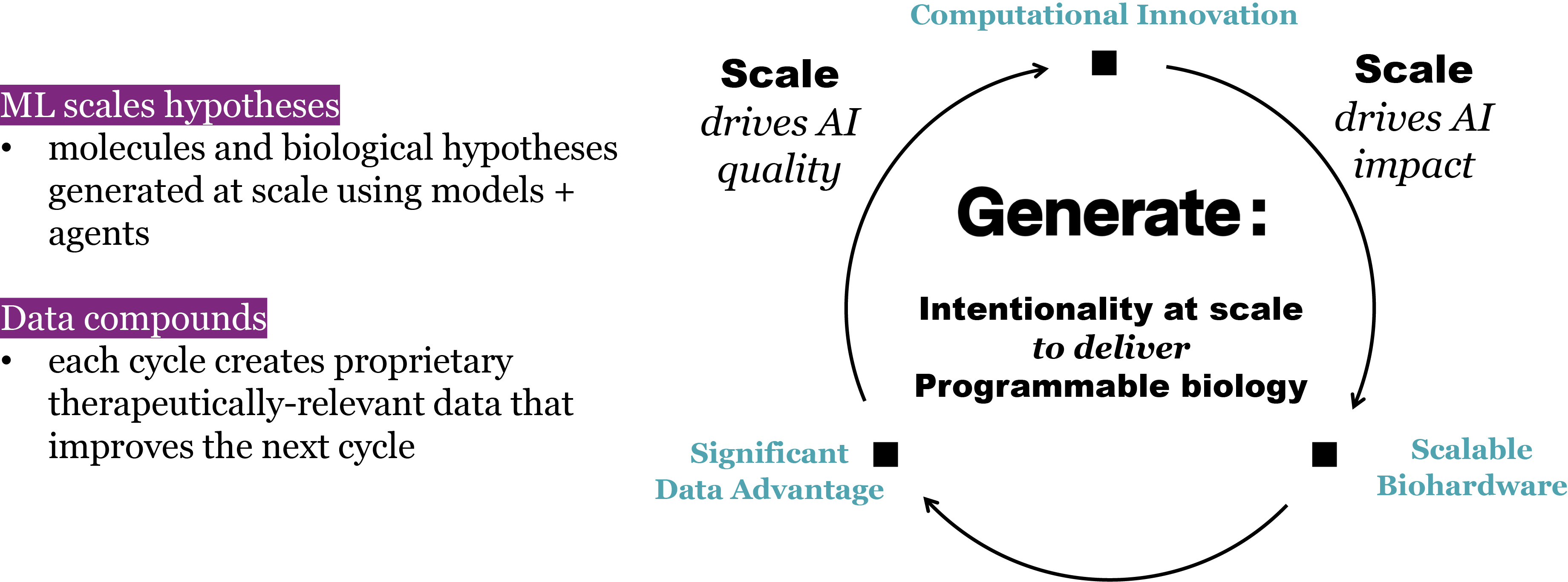
at time t to predicted denoised coordinates 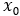 . B. We parameterize this in terms of a random graph neural network with long-range connectivity inspired by efficient N-body algorithms (middle) and a fast method for solving for a global consensus structure given predicted inter-residue geometries (right). Another graph-based design network (A, top right) generates protein sequences and side-chain conformations conditionally based on the sampled backbone. C. The time-dependent protein prior learnt by the diffusion model can be combined with composable restraints and constraints for the programmable generation of protein systems.
. B. We parameterize this in terms of a random graph neural network with long-range connectivity inspired by efficient N-body algorithms (middle) and a fast method for solving for a global consensus structure given predicted inter-residue geometries (right). Another graph-based design network (A, top right) generates protein sequences and side-chain conformations conditionally based on the sampled backbone. C. The time-dependent protein prior learnt by the diffusion model can be combined with composable restraints and constraints for the programmable generation of protein systems.