UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31 , 2025
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM TO
Commission File Number 001-42437
(Exact name of Registrant as specified in its Charter)
| | | |
| (State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
| | | |
| (Address of principal executive offices) | | (Zip Code) |
Registrant’s telephone number, including area code: +61 7 3152 3200
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| | | The |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
YES ☐ ☒
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act.
YES ☐ ☒
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
☒ NO ☐
Indicate by check mark whether the Registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit such files).
☒ NO ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| | ☒ | Smaller reporting company | |
| Emerging growth company | | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
YES ☐ NO ☒
The registrant was no t a public company as of the last business day of its most recently completed second fiscal quarter and, therefore, cannot calculate the aggregate market value of its voting equity held by non-affiliates as of such date.
The number of shares of Registrant’s Common Stock outstanding as of February 25, 2026 was 97,232,054 .
| TABLE OF CONTENTS | |||
| Page | |||
| 6 | |||
|
Item 1.
|
6 | ||
|
Item 1A.
|
27 | ||
|
Item 1B.
|
56 | ||
|
Item 1C.
|
56 | ||
|
Item 2.
|
57 | ||
|
Item 3.
|
57 | ||
|
Item 4.
|
57 | ||
| 58 | |||
|
Item 5.
|
58 | ||
|
Item 6.
|
58 | ||
|
Item 7.
|
59 | ||
|
Item 7A.
|
66 | ||
|
Item 8.
|
67 | ||
|
Item 9.
|
107 | ||
|
Item 9A.
|
107 | ||
|
Item 9B.
|
108 | ||
|
Item 9C.
|
108 | ||
| 108 | |||
|
Item 10.
|
108 | ||
|
Item 11.
|
115 | ||
|
Item 12.
|
123 | ||
|
Item 13.
|
129 | ||
|
Item 14.
|
132 | ||
| 132 | |||
|
Item 15.
|
132 | ||
|
Item 16.
|
134 | ||
INTRODUCTION
Prior to the consummation of our initial public offering in December 2024, we completed a series of reorganization transactions (the “Reorganization”). Unless otherwise indicated or context otherwise requires in this Annual Report on Form 10-K (this “Form 10-K”), all references in this Form 10-K to the “Company,” “Anteris,” “Anteris®,” “we,” “us” and “our” refer to Anteris Technologies Pty Ltd (“ATPL”, formerly Anteris Technologies Ltd) prior to the Reorganization and Anteris Technologies Global Corp. (“ATGC”) after the Reorganization, and for purposes of this Form 10-K:
•
“ADAPT® anti-calcification tissue” refers to the tissue produced by the ADAPT® tissue engineering process, which transforms xenograft tissue (bovine heart tissue) into a durable bioscaffold which Anteris uses in its DurAVR® THV to mimic human tissue in aortic valve replacement.
•
“Aldehydes” refers to organic compounds.
•
“Aortic stenosis” refers to the narrowing of the aortic valve restricting the flow of blood from the left ventricle (lower chamber of the heart) to the aorta (main artery).
•
“Bioscaffold” refers to a durable structure engineered from biological material.
•
“Coaptation” refers to the portion of the leaflets that touch when the aortic valve is in the closed position.
•
“ComASUR® Delivery System” refers to the balloon expandable system which provides controlled deployment and accurate placement of the DurAVR® THV, designed to achieve precise alignment with the heart’s native commissures to achieve ideal valve positioning.
•
“Commissure alignment” refers to the position of the transcatheter aortic valve replacement leaflets in line with the anatomical orientation of the recipient’s native valve leaflets.
•
“Commissures” refers to where the valve leaflets are attached to the aortic wall inside the aortic sinus of Valsalva.
•
“Cytotoxicity” refers to toxicity to cells.
•
“Doppler velocity index” and “DVI” refer to the index that expresses the EOA as a proportion of valve area, with DVI representing the physical ratio of a patient’s aortic valve area to the left ventricular outflow tract area. A higher DVI indicates improved blood flow through the aortic valve. DVI is independent of the flow state (like gradient) and diameter (like EOA).
•
“DurAVR® THV” refers to a transcatheter heart valve (“THV”) developed by Anteris. It is a novel, biomimetic (meaning human-like) valve made from a single-piece of native-shaped ADAPT® tissue and is used for the treatment of aortic stenosis. The DurAVR® THV (new aortic valve) is placed within the diseased aortic valve via a minimally invasive procedure.
•
“Effective orifice area” and “EOA” refer to the smallest cross-sectional area of the aortic valve opening that is available for blood flow. A larger EOA reduces the work the left ventricle (heart chamber) must do to pump blood through the valve. Patients with severe aortic stenosis typically have an EOA of ≤ 1 cm2.
•
“Exercise capacity” refers to a measure of a patient’s exercise ability, measured in clinical trials by a six minute walk test (“6MWT”), which scores a person on the distance they can cover in six minutes of walking.
•
“Flow displacement” and “FD” refer to a marker of flow eccentricity in the ascending aortic root. Flow in the ascending aortic root is mainly laminar with a flow displacement ranging from 6 – 15% only. A higher degree of FD reflects abnormal turbulent flow.
•
“Flow reversal ratio” or “FRR” is calculated at peak systole in the ascending aorta. At this point there should be almost no backward flow, and any backward flow is considered abnormal. FRR represents the ratio of backward and forward flow at peak systole.
•
“Hemodynamics” refers to how blood flows through the blood vessels.
•
“Laminar flow” refers to a smooth, streamlined flow of blood. In a healthy heart, aortic flow is predominantly laminar during systole (when the left ventricle contracts and pumps blood into the aorta). Abnormal aortic flow is associated with turbulence, which can increase the risk of morbidity and increase the stress on the valve leaflets leading to increased wear and tear and subsequent structural valve deterioration.
•
“Mean pressure gradient” and “MPG” refer to the average pressure across the aortic valve between the left ventricle and aorta. Patients with severe aortic stenosis have MPG ≥ 40 mmHg. Post-TAVR MPG is expected to decrease, which indicates that the left ventricle is not working as hard to pump blood through the aortic valve.
•
“Transcatheter aortic valve replacement” or “TAVR” refer to a minimally invasive procedure for the treatment of aortic stenosis. A new aortic valve is placed inside the diseased valve, meaning the old, damaged valve is not removed.
•
“ViV” refers to valve-in-valve.
•
“Xenograft” refers to a tissue that is derived from a species that is different from the recipient of the specimen, meaning tissue from animal species.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
All statements in this Form 10-K, other than statements of historical facts, including statements regarding our future results of operations and financial position, business strategy, product development, and plans and objectives of management for future operations, are forward-looking statements. These forward-looking statements generally are identified by the words “believe,” “project,” “expect,” “anticipate,” “estimate,” “intend,” “budget,” “target,” “aim,” “strategy,” “plan,” “guidance,” “outlook,” “may,” “should,” “could,” “will,” “would,” “will be,” “will continue,” “will likely result” and similar expressions, although not all forward-looking statements contain these identifying words. Forward-looking statements, which are subject to risks, include, but are not limited to, statements about:
•
our current and future research and development (“R&D”) activities, including clinical testing and manufacturing and related costs and timing;
•
our product development and business strategy, including the potential size of the markets for our products and future development and/or expansion of our products in our markets;
•
our ability to commercialize products and generate product revenues;
•
any statements concerning anticipated regulatory activities, including our ability to obtain regulatory clearances;
•
our R&D expenses;
•
risks facing our operations and intellectual property;
•
sufficiency of our capital resources; and
•
our ability to raise additional funding when needed.
We have based the forward-looking statements contained in this Form 10-K largely on our current expectations, estimates, forecasts and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. In light of the significant uncertainties in these forward-looking statements, you should not rely upon forward-looking statements as predictions of future events. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Form 10-K, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur at all. You should refer to the section titled “Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material.
The forward-looking statements made in this Form 10-K relate only to events as of the date on which the statements are made. Except as required by law, we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise. The Private Securities Litigation Reform Act of 1995 and Section 27A of the Securities Act of 1933 (the “Securities Act”), do not protect any forward-looking statements that we make within this Form 10-K.
You should read this Form 10-K and the documents that we reference in this Form 10-K completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements in this Form 10-K by these cautionary statements.
This Form 10-K contains certain data and information that we obtained from various publications, including industry data and information from Future Market Insights, Inc. (“FMI”). Statistical data in these publications also include projections based on a number of assumptions. The global, North American and European TAVR markets may not grow at the rate projected by market data or at all. Failure of the global, North American and European TAVR markets to grow at the projected rate may have a material and adverse effect on our business and the market price of our Common Stock, par value $0.0001 per share (“Common Stock”), and CHESS Depository Interests (“CDIs”). All references in this Form 10-K to Common Stock shall include the shares represented by CDIs unless the context suggests otherwise. In addition, the nature of the medical technology industry results in significant uncertainties for any projections or estimates relating to the growth prospects or future condition of our industry. Furthermore, if any one or more of the assumptions underlying the market data are later found to be incorrect, actual results may differ from the projections based on these assumptions. You should not place undue reliance on these forward-looking statements.
CAUTIONARY NOTE REGARDING INDUSTRY AND MARKET DATA
This Form 10-K includes information concerning the Company’s industry and the markets in which it operates that is based on information from various sources including public filings, internal company sources, various third-party sources and management estimates. In addition, this Form 10-K contains information from a report prepared by FMI, a market research firm that we commissioned to provide information on the global transcatheter heart valve replacement market. Management estimates regarding the Company’s position, share and industry size are derived from publicly available information and its internal research and are based on a number of key assumptions made upon reviewing such data and the Company’s knowledge of such industry and markets, which it believes to be reasonable. In some cases, we do not expressly refer to the sources from which this information is derived. While the Company believes the industry, market and competitive position data included in this Form 10-K is reliable and is based on reasonable assumptions, such data is necessarily subject to a high degree of uncertainty and risk and is subject to change due to a variety of factors, including those described in sections titled “Special Note Regarding Forward-Looking Statements,” “Risk Factors” and elsewhere in this Form 10-K. These and other factors could cause results to differ materially from those expressed in the estimates included in this Form 10-K. The Company has not independently verified any data obtained from third-party sources and cannot assure you of the accuracy or completeness of such data.
RISK FACTOR SUMMARY
Investing in shares of our Common Stock involves a high degree of risk. You should carefully consider the following risks and uncertainties, together with all of the other information contained in this Form 10-K, including the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our audited consolidated financial statements and related notes included elsewhere in this Form 10-K, together with our other publicly available filings with the Securities and Exchange Commission (the “SEC”). The occurrence of any of the following risks, or of additional risks and uncertainties not presently known to us or that we currently believe not to be material, could materially and adversely affect our business, financial condition, reputation, and results of operations. Set forth below is a summary of some, but not all, of the principal risks we face:
•
We have a history of operating losses and may not achieve or maintain profitability in the future.
•
Unsuccessful clinical trials or procedures relating to our products could have a material adverse effect on our prospects.
•
If we are unable to successfully identify, develop, obtain and maintain regulatory clearance or approval for and ultimately commercialize any of our current or future products, or experience significant delays in doing so, our business, financial condition and results of operations will be materially adversely affected.
•
Even if a product receives regulatory clearance or approval, it may still face development and regulatory difficulties that could delay or impair future sales of products.
•
Even with regulatory clearance or approval to bring a product to market, our profitability may be impacted by ongoing coverage and reimbursement determinations by government health care programs and other third-party payors for our products, or procedures and services that rely on our products.
•
Our products are in development and may not achieve market acceptance, if approved, which could limit our growth and adversely affect our business, financial condition, and results of operations.
•
We may find it difficult to enroll patients in our clinical trials, and patients could discontinue their participation in clinical trials, which could delay or prevent clinical trials and make those trials more expensive to undertake.
•
We operate in a highly competitive and rapidly changing industry, and if we do not compete effectively, our business will be harmed.
•
The success of many of our products may depend upon the knowledge and experience of certain key physicians and heart valve centers.
•
We rely on third parties to conduct our clinical trials and preclinical studies. If these third parties do not successfully carry out their contractual duties, comply with applicable regulatory requirements or meet expected deadlines, our development programs and our ability to seek or obtain regulatory clearance and approval for or commercialize our products may be delayed.
•
We are subject to various risks relating to international activities that could affect our profitability, including risks associated with currency fluctuations and changes in foreign currency exchange rates.
•
Any failure to protect our information technology infrastructure and our products against cyber-based attacks, network security breaches, service interruptions, artificial intelligence or data corruption could materially disrupt our operations and adversely affect our business and operating results.
•
Artificial intelligence technologies could present business, compliance and reputational risks.
•
Increased emphasis on environmental, social, and governance matters may have an adverse effect on our business, financial condition, results of operations and reputation.
•
We could become exposed to product liability claims that could harm our business, and we may be unable to obtain insurance coverage at acceptable costs and adequate levels.
•
Use of our products in unapproved circumstances could expose us to liabilities.
•
Our products and operations are subject to extensive government regulation, including environmental, health and safety regulations, which could result in substantial costs. Furthermore, any failure to comply with applicable requirements could harm our business.
•
Healthcare policy changes may have a material adverse effect on us.
•
We could be exposed to significant liability claims if we are unable to obtain insurance at acceptable costs and adequate levels or otherwise protect ourselves against potential product liability claims.
•
Tax laws, regulations, and enforcement practices are evolving and may have a material adverse effect on our results of operations, cash flows and financial position.
•
Our success depends on our ability to protect our intellectual property and our proprietary technology.
•
Intellectual property rights of third parties could adversely affect our ability to commercialize our products.
•
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
•
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
•
Any difficulty with protecting our intellectual property could diminish the value of our intellectual property rights in the relevant jurisdiction.
•
Medtronic plc (“Medtronic”) beneficially owns a significant equity interest in us and its interests may conflict with our or your interests.
•
We will require substantial additional future financing and, until commercialization of our products, our cash burn in future periods may be higher than anticipated. We may be unable to raise sufficient capital in future financings, including to account for unanticipated cash burn, which could have a material impact on our R&D programs or commercialization of our products.
•
Our Common Stock is listed on The Nasdaq Global Market (“Nasdaq”), but the market price and trading volume may continue to fluctuate and remain limited in terms of liquidity.
•
Our Second Amended and Restated Certificate of Incorporation (“Charter”) and Amended and Restated Bylaws (“Bylaws”) contain anti-takeover provisions that could delay or discourage takeover attempts that stockholders may consider favorable.
Overview
Anteris is a structural heart company dedicated to revolutionizing cardiac care by pioneering science-driven and measurable advancements to restore heart valve patients to healthy function. Our lead product, the DurAVR® THV System, was designed in collaboration with the world’s leading interventional cardiologists and cardiac surgeons to treat aortic stenosis — a potentially life-threatening condition resulting from a narrowing of the aortic valve. The balloon-expandable DurAVR® THV is the first biomimetic valve, which is shaped to mimic the performance of a healthy human aortic valve and aims to replicate normal aortic blood flow. Our DurAVR® THV System consists of a single-piece, biomimetic valve made with our proprietary ADAPT® tissue-enhancing technology and deployed with our balloon expandable ComASUR® Delivery System. ADAPT® is our proprietary anti-calcification tissue shaping technology that is designed to reengineer xenograft tissue into a pure, single-piece collagen bioscaffold. Our patented ADAPT® tissue has been clinically demonstrated to be calcium free for up to 10 years post-procedure, according to Performance of the ADAPT-Treated CardioCel® Scaffold in Pediatric Patients With Congenital Cardiac Anomalies: Medium to Long-Term Outcomes, published by William Neethling et. al., and has been distributed for use in over 55,000 patients globally in other indications. Our balloon expandable ComASUR® Delivery System, which was developed in consultation with physicians, is designed to provide precise alignment with the heart’s native commissures to achieve accurate placement of the DurAVR® THV. As of December 2025, more than 130 patients have been implanted with the DurAVR® THV worldwide.
Aortic stenosis is one of the most common and serious valvular heart diseases. It is fatal in approximately 50% of patients if left untreated after two years, and no pharmacotherapy is available to treat this disease. Aortic stenosis causes a narrowing of the heart’s aortic valve, which reduces or blocks the amount of blood flowing from the heart to the body’s largest artery, the aorta, and from there to the rest of the body. Minimally-invasive transcatheter aortic valve replacement (“TAVR”), which the U.S. Food and Drug Administration (“FDA”) initially approved in 2011 for high surgical risk patients, has emerged as an alternative to open-heart surgery. In 2019, the FDA also approved TAVR for use in low-risk surgical patients. These low-risk surgical patients are often younger persons within the geriatric population that require heart valves with longer durability and pre-disease hemodynamics for an improved quality of life. More generally, patients with aortic valve stenosis are now being diagnosed at a younger age. Yet, according to a publication in The Journal of American Medical Association, only 15-20% of severe aortic stenosis cases are treated today.
While previous generations of TAVRs were designed for older, high risk patients, our DurAVR® THV System is designed to be a solution for all patients, including both older, younger and less-active patients. Our first in class DurAVR® THV is a single-piece valve with a novel, biomimetic design that aims to replicate the normal blood flow of a healthy human aortic valve as compared to traditional three-piece aortic valves. The DurAVR® THV System has shown restoration of laminar flow similar to individuals with a healthy aortic valve, and early left ventricular reverse remodeling compared with pre-TAVR and similar to healthy controls.
In a pooled analysis of 100 patients derived from our ongoing First-In-Human (“FIH”) study (referred to as the “EMBARK” study) and early feasibility studies (“EFS”) conducted in the United States and Europe, the DurAVR® THV demonstrated single digit mean gradients, large effective orifice areas (“EOAs”), no moderate or severe paravalvular leaks and no valve related mortality, with 97% freedom from prosthesis-patient mismatch (“PPM”). These results were observed in a cohort of small annuli patients similar to the one reported in the SMall Annuli Randomized To Evolut™ or SAPIEN™ Trial (“SMART”), a prospective, multicenter, randomized controlled trial evaluating transcatheter aortic valve performance in patients with small aortic annuli. PPM affects a significant proportion of TAVR patients, particularly patients with a small aortic annulus and has been associated with impaired long-term survival following surgical aortic valve replacement (“SAVR”).
In addition, our DurAVR® THV has been developed with the aim to increase durability and last longer than traditional three-piece designs through the use of our ADAPT® anti-calcification tissue including a molded single-piece of tissue designed to mimic the performance of a pre-disease human aortic valve, which we believe can result in improved hemodynamics as compared to traditional three-piece designs. These designs and features cumulatively aim to provide a better quality of life as compared to the current standard of care associated with traditional three-piece designs. We intend to test these features in the DurAVR® THV randomized, global pivotal study (the “PARADIGM Trial”).
The PARADIGM Trial is a prospective, randomized, controlled multicenter, international study wherein subjects are randomized to receive either a TAVR using the DurAVR® THV or TAVR using a commercially available and approved THV in an ‘All Comers Randomized Cohort.’ The primary end point of the PARADIGM Trial is a composite of all-cause mortality, all stroke and cardiovascular hospitalizations at 1-year post-procedure, according to Valve Academic Research Consortium‑3 (“VARC-3”), a globally accepted consensus framework that standardizes clinical endpoint definitions for aortic valve clinical research. The endpoint will be evaluated as a non-inferiority analysis. We anticipate that the subjects will include a broad array of risk profiles. Subjects with a failed surgical bioprosthesis in need of a ViV TAVR are enrolled in a separate parallel registry.
In 2025, we advanced regulatory activities in Europe, with the goal of securing approval to commence the PARADIGM Trial in a number of European countries. In October 2025, we secured the first European regulatory approval in Denmark and subsequently enrolled and treated the first patients marking the formal initiation of the PARADIGM Trial. In November 2025, we also received Investigational Device Exemption (“IDE”) approval from the FDA for the PARADIGM Trial. The FDA granted a staged approval authorizing enrollment of the first 200 patients. We may request authorization to expand enrollment for the remaining subjects through an IDE supplement. Throughout the year, our cross‑functional teams continued to execute site activation, regulatory preparation, and operational readiness activities in anticipation of regulatory approval in each participating country.
We expect the data from the PARADIGM Trial could provide the clinical evidence required for regulators to approve commercialization. This includes premarket approval that is required for commercialization of the DurAVR® THV System in the United States and CE Mark approval in Europe.
We continued strengthening our operational infrastructure during the year, advancing quality management system buildout to support upcoming clinical activities and future ISO 13485 certification. Key quality procedures and standard operating documents were released to establish the framework for a mature, compliant system and mitigate audit risk. In parallel, manufacturing scale-up activities progressed, including cross-training of inspection personnel, expansion of clean room capacity, and ongoing process development initiatives to ensure robust, high-yield production in line with projected demand.
We are a development stage company and have incurred net losses each year since operation, however, we believe that we have significant growth potential in a large, underpenetrated and growing TAVR market. Since the inception of the TAVR procedure, the annual volume of TAVR procedures in the United States has increased significantly year-over-year, with an estimated 73,000 patients having undergone a TAVR procedure in the United States in 2019 according to the STS/ACC TVT Registry. According to FMI, a market research firm, the total global market opportunity for TAVR in relation to severe aortic stenosis and in relation to ViV procedures is expected to reach $9.9 billion and $2.5 billion, respectively, in 2028.
Our innovation-focused R&D practice is driven by rapid technological advancement and significant input from leading interventional cardiologists and cardiac surgeons. As a company that is primarily in the development phase, we currently generate small amounts of revenue and income which are insufficient to cover our investment in research, development and operational activities resulting in recurring net operating losses, incurred since inception. We, like other development stage medical device companies, experience challenges in implementing our business strategy due to limited resources and a smaller capital base as we prioritize product development, minimize the period to the commencement of commercial sales, ensure our focus on quality as well as scale our operations. The development and commercialization of new medical devices is highly competitive. Those competitors may have substantial market share, substantially greater capital resources and established relationships with the structural heart community, potentially creating barriers to adoption of our technology. Our success will partly be based on our ability to educate the market about the benefits of our disruptive technology including current unmet clinical needs compared to commercially available devices as well as how we plan to capture market share post commercialization.
We are dedicated to developing technological enhancements and new indications for existing products, and less invasive and novel technologies to address unmet patient needs in structural heart disease. That dedication leads to our initiation and participation in clinical trials that seek to prove our pipeline is safe and effective as the demand for clinical and economic evidence remains high.
From time to time, we enter into strategic agreements aimed at enhancing our business operations and profitability. For example, in April 2023, we invested in and entered into a development agreement (the “Development Agreement”) with, v2vmedtech, inc. (“v2vmedtech”), which develops an innovative heart valve repair device for the minimally invasive treatment of mitral and tricuspid valve regurgitation.
Market Opportunity
According to the World Bank, the total population over 65 in the United States and the European Union was approximately 173.6 million as of 2024. According to FMI, the total global market opportunity for TAVR in relation to severe aortic stenosis and in relation to ViV procedures is expected to reach $9.9 billion and $2.5 billion, respectively, in 2028. The key specific markets that our Company is initially targeting are North America and Europe due to these markets accounting for the majority of the above global opportunity. FMI indicated that the North American and European markets averaged 53% and 38% of the global market share, respectively, during the period 2016 to 2023. FMI forecasts that the market opportunity in relation to severe aortic stenosis for North America and Europe will reach $5.5 billion and $3.7 billion, respectively, in 2028; and the market opportunity in relation to ViV procedures is forecast to reach $1.5 billion and $0.8 billion, respectively, in 2028. To calculate these future market values, FMI has relied on actual data from 2023 collated from a variety of published sources and key medical experts and applied a projected CAGR of 14.9% for the global market, 16.2% for the North American market, and 14.0% for the European market. A non-exhaustive list of factors that may impact these forecast calculations include key players’ historic growth; companies and manufacturers working together to develop new, affordable and timesaving technologies; new product launches and approvals; rising demand for THV replacement; availability and cost of products; growing investment in healthcare expenditure; and increased regulatory focus on patient safety and reimbursement policies. In addition, we expect the TAVR market to benefit from general trends, including an aging population, earlier diagnosis of aortic stenosis, increased incidence of obesity and diabetes (which contribute to heart disease), as well as the broader patient populations’ desire to pursue a more active lifestyle.
Since the inception of the TAVR procedure, the annual volume of TAVR procedures in the United States has increased significantly year-over-year, with an estimate of nearly 100,000 patients having undergone a TAVR procedure in the United States in 2022 according to the TVT Registry. We believe that the rising geriatric population and the growing cardiovascular device market provides us with a clear business opportunity. The use of healthcare services is significantly higher among older people.
DurAVR® THV’s single-piece native shaped biomimetic design replicates the performance of a healthy human aortic valve and is designed to restore normal blood flow as compared to traditional three-piece transcatheter valves, either balloon expandable or self-expanding, which do not restore normal aortic flow. We believe this design, in combination with the ADAPT® tissue technology, has the potential to allow the DurAVR® THV to last longer than traditional three-piece aortic valves, which have multiple leaflets sewn together that may lead to compromised durability.
Our Product Candidates
DurAVR® THV, which employs our ADAPT® anti-calcification tissue and is deployed using our ComASUR® Delivery System, is currently in clinical development.

DurAVR® Transcatheter Heart Valve System
Our DurAVR® THV is a novel transcatheter aortic valve designed in collaboration with the world’s leading interventional cardiologists and cardiac surgeons to treat symptomatic severe aortic stenosis – a potentially life-threatening condition resulting from the narrowing of the aortic valve. The balloon-expandable DurAVR® THV is the first biomimetic valve, shaped to mimic the performance of a healthy human aortic valve and aims to replicate normal aortic blood flow. Our DurAVR® THV is made using a single piece of molded bovine pericardial tissue treated with ADAPT®, our patented anti-calcification tissue technology, sutured inside a cobalt-chromium frame. ADAPT® tissue, which is FDA-cleared, has been used clinically for over 10 years and distributed for use in over 55,000 patients worldwide. The DurAVR® THV System is comprised of the DurAVR® valve, the ADAPT® tissue, and the balloon-expandable ComASUR® Delivery System. These designs and features cumulatively aim to restore a better quality of life compared to the current standard of care associated with traditional three-piece designs. We intend to evaluate the safety and efficacy of the DurAVR® THV against commercially approved TAVR devices in the global pivotal PARADIGM Trial (NCT07194265).
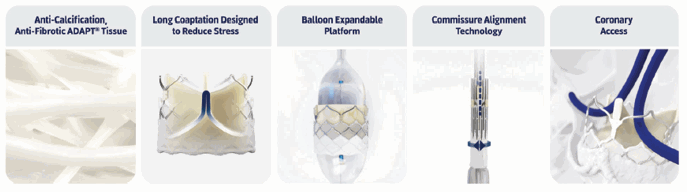
The DurAVR® THV has the following attributes:
•
it is the first transcatheter aortic valve to use a patented construction of a molded single-piece of bioengineered tissue (our ADAPT® anti-calcification tissue with molded leaflets (see “ADAPT® Anti-Calcification Tissue”));
•
it has fewer sutures and seams when compared with conventional valves, thereby preserving tissue integrity with the intent to reduce calcification risk to extend valve durability;
•
it is uniquely shaped to emulate the performance of a healthy human valve and produce long leaflet coaptation, laminar flow and near-normal hemodynamics;
•
it has large open cells in the stent frame to facilitate coronary access; and
•
it utilizes the ComASUR® balloon expandable Delivery System (see “ComASUR® Delivery System”) for controlled deployment and accurate placement.
ADAPT® Anti-Calcification Tissue
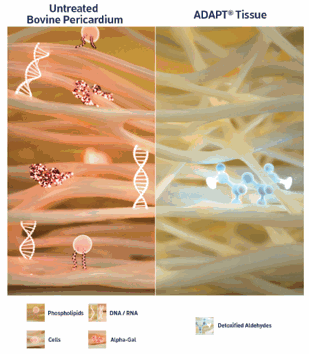
The ADAPT® tissue engineering process is an anti-calcification preparation that transforms xenograft tissue (bovine pericardium) into durable bioscaffolds that are used to mimic human tissue for surgical repair in multiple settings, including aortic valve replacement. The outcome of the ADAPT® tissue engineering process is a novel, acellular, biostable and non-calcifying biomaterial.
The ADAPT® tissue engineering process involves multiple steps to transform bovine pericardium into a durable bioprosthetic material. Bovine spongiform encephalopathy-free bovine pericardium is decellularized to remove all cellular antigens that initiate an immune response. The material is then crosslinked to enable maintenance and stabilization of strength and elasticity to improve mechanical resistance. The cytotoxicity is further reduced using detoxification and sterilization processes and anti-calcification methodology to remove and bind aldehydes and enable safe storage in a non-glutaraldehyde solution. Post-implantation, ADAPT® tissue provides a scaffold for cell migration to create the optimal environment. Migrated cells can stimulate site-specific remodeling and repair and enable the formation of new blood vessels.
Our proprietary ADAPT® tissue has been clinically demonstrated to be calcium-free for up to 10 years post-procedure, according to Performance of the ADAPT-Treated CardioCel® Scaffold in Pediatric Patients With Congenital Cardiac Anomalies: Medium to Long-Term Outcomes, published by William Neethling et. al., and it has been distributed for use in over 55,000 patients globally in other indications. Our ComASUR® balloon-expandable Delivery System, which was developed in consultation with physicians, is designed to provide precise alignment with the heart’s native commissures to achieve accurate placement of the DurAVR® THV.
To meet the need for a durable THV, made from ADAPT® tissue scaffold, we have created the DurAVR® THV, which is a first in class, biomimetic single-piece valve with optimal hemodynamic and durability properties. Based on published clinical data in several peer-reviewed journals, including The Journal of Thoracic and Cardiovascular Surgery, the Expert Review of Medical Devices, and Interactive Cardiovascular and Thoracic Surgery, ADAPT® has been observed to offer potentially significant improvements compared with other widely available commercial processes adopted by healthcare providers, including with respect to bio-compatibility, durability, strength, pliability, functionality and controlled remodeling.
ComASUR® Delivery System
Our ComASUR® Delivery System is a physician-developed balloon expandable delivery system that contains a reinforced steerable catheter for a precise deflection through the heart anatomy in a controlled manner to avoid damage to the aorta. This delivery system provides controlled deployment and accurate placement of our DurAVR® THV. Our ComASUR® Delivery System is designed to achieve precise alignment with the heart’s native commissures to achieve ideal valve positioning.
Within the ComASUR® Delivery System, we have rotational control of the DurAVR® valve with the native commissures. This allows for commissure alignment, which is not achieved consistently in competitive delivery systems. Commissure alignment is important because it positions the transcatheter valve leaflets in line with the patient’s native anatomy, supporting optimal hemodynamics, facilitating future coronary access, and enabling consistent device performance. This feature positions the TAVR valve leaflets exactly in line with the anatomical orientation of the recipient’s native valve leaflets. We have a patent pending for this system.
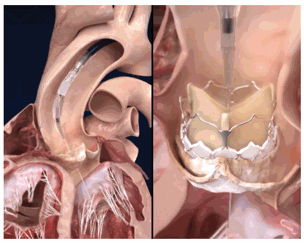
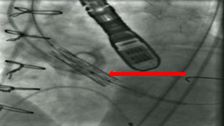
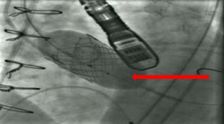
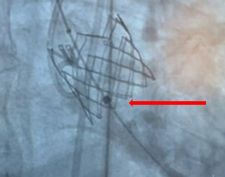
The ComASUR® Delivery System provides even balloon expansion for the accurate placement of the DurAVR® THV as well as ease of use. Under fluoroscopic guidance the physician precisely aligns the DurAVR® THV with the native annulus before deployment in the following manner:
|
First, the balloon starts out as collapsed.
|
|
|
|
|
The balloon is then expanded and the DurAVR® THV is deployed.
|
|
|
|
|
Finally, the balloon is deflated and removed.
|
Clinical Results and Trials
Over 130 patients have been successfully treated with the DurAVR® THV, including de novo (first time) aortic stenosis cases, ViV patients and complex anatomies such as bicuspid aortic valve patients. We continue to expand the level of global experience and build the body of clinical evidence for the DurAVR® THV System through our clinical development program, comprised of the ongoing EMBARK study, EFSs in the United States and European Union (the “U.S. EFS” and “EU EFS,” respectively) and the recently initiated global, pivotal PARADIGM Trial, in addition to compassionate use cases.
The EMBARK study, initiated in November 2021 in Tbilisi, Georgia, has enrolled nine cohorts and demonstrated consistent device performance, favorable hemodynamic outcomes, and an acceptable safety profile through available follow-up periods. In addition, we enrolled 15 patients across four centers in our U.S. EFS, achieving 100% implant success with no mortality, disabling stroke, or life-threatening bleeding at 30 days. Our EU EFS enrolled 15 patients to further evaluate safety and ViV performance in a controlled setting. 30-day data from these studies were pooled and recently published in EuroIntervention. This data demonstrates promising clinical and echocardiographic outcomes in patients with small aortic annuli, a population at greater risk for impaired valve hemodynamics. The overall technical success rate was high at 93% with no deaths, a low stroke rate (2%), single-digit gradients (8.2 mmHg), large EOAs (2.2 cm2), and low rate of moderate or greater prosthesis-patient mismatch (3%) through 30 days.
Building on these clinical programs, in 2025 we initiated our PARADIGM Trial, a global, prospective, randomized, controlled, multicenter study comparing DurAVR® THV to commercially available THVs. The PARADIGM Trial is designed to generate the clinical evidence necessary to support global regulatory approval. The first patients were enrolled in October 2025, and in November 2025, we announced that the FDA granted IDE approval for a staged enrollment for the first 200 patients in the PARADIGM Trial in the United States.
Competition
We compete in the cardiovascular device market, and in particular the TAVR market. These markets are characterized by rapid change resulting from technological advances, innovations and scientific discoveries. Our products face a mix of competitors ranging from large manufacturers with multiple business lines to small manufacturers offering a limited selection of products. In addition, we face competition from providers of other medical therapies, such as pharmaceutical companies. Our primary competitors include Edwards Lifesciences Corporation and Medtronic. Currently, no competitor has a single-piece tissue TAVR commercially available or has publicly disclosed that a single-piece tissue TAVR is in development.
Major shifts in industry market share have occurred in connection with product corrective actions, physician advisories, safety alerts, results of clinical trials to support superiority claims, and publications about products, reflecting the importance of product quality, product efficacy and quality systems in the medical technology industry. In the current environment of managed care, economically motivated customers, consolidation among healthcare providers, increased competition, declining reimbursement rates, and national and provincial tender pricing, competitively priced product offerings are essential to our business. In order to compete effectively, we must continue to create or acquire advanced technology, incorporate this technology into proprietary products, obtain regulatory approvals in a timely manner, maintain high-quality manufacturing processes, and successfully market these products.
Intellectual Property
We rely on a combination of patent, copyright, trademark and trade secret laws and confidentiality and invention assignment agreements to protect our intellectual property rights in the United States and other markets. United States federal registrations for trademarks can remain in force in perpetuity, provided the mark is still being used in commerce and the maintenance/renewal filings are made as required by the sixth year after registration, by the tenth year after registration, and every ten years thereafter.
As of December 31, 2025, Anteris owned a total of 47 active patents expiring between 2032 and 2045, and 75 pending patent applications, as further detailed below.
In the category of prosthetic heart valve devices, we are the sole owner of eight active United States patents, seven pending United States patent applications, seven active Australian patents, three pending Australian patent applications, eight active patents in other countries, and 31 pending applications in other countries. These patents and pending applications are directed to features that are expected to provide competitive advantages such as: a novel process for production of calcification resistant cross-linked biomaterials for the prosthetic valve; three-dimensional molded heart valve leaflets made of cross-linked biomaterial that mimic the performance of a native heart valve designed to provide enhanced performance characteristics such as low mean pressure gradient, low leaflet stress, large open area, high coaptation area and high duration in an open state, to name a few; a prosthetic heart valve that has localized protective covering members that prevent direct contact between the valve and the stent frame to enhance the durability and longevity of the prosthetic valve when the valve is in an open state; and attachment of the biomaterial valve to the stent frame in a novel manner that reduces stresses on the biomaterial of the prosthetic valve.
In the category of delivery systems for the prosthetic heart valve devices, we are the sole owner of three active United States patents, seven pending United States patent applications, one active Australian patent, three pending Australian patent applications, two pending PCT applications, one active patent in other countries, and 15 pending applications in other countries. These patents and pending applications are directed to features that are expected to provide competitive advantages such as: controllable and predictable commissural alignment; a balloon folding technique that mitigates valve rotations during expansion; a single-use valve crimping device; and a delivery catheter hard stop member made of a braided metal material that provides improved trackability, effective expansion of the delivery sheath during advancement, and increased longitudinal compressive strength that serves to maintain the longitudinal position of the prosthetic heart valve on the balloon member.
In the category of sterilization and storage of the prosthetic heart valve devices, we are the sole owner of two active United States patents, one active Australian patent, seven active patents in other countries, and one pending application in other countries. These patents and pending applications are directed to features that are expected to provide competitive advantages such as a novel process for sterilizing the valve made of collagen-containing implantable biomaterials and storage thereafter.
In the category of packaging, we are the sole owners of two active United States patents, one pending United States patent application, two active Australian patents, one pending Australian patent application, five active patents in other countries, and four pending patent applications in other countries. These patents and pending applications are directed to features that are expected to provide competitive advantages such as a packaging design that includes integrated components and mechanisms for preparing and mounting the valve on the delivery catheter system to make the clinician’s valve preparation process more efficient and user-friendly.
Anteris holds a 30% interest in v2vmedtech. v2vmedtech’s intellectual property is directed to implantable medical devices for mitigating heart valve regurgitation. Using a transcatheter deployment technique, one or more clip devices are attached to the leaflets of a patient’s mitral or tricuspid heart valve to permanently join together edge portions of the leaflets. This is often referred to as an edge-to-edge repair procedure. As of December 31, 2025, v2vmedtech had 13 pending patent applications and is the exclusive licensee of two pending patent applications owned by Columbia University.
We have trademark registrations for several of our most material marks, including “ADAPT,” “ADAPT FOR LIFE”, “ANTERIS”, “ComASUR”, “DurAVR”, and “GYNECEL”. Our filing for the “ANTERIS” trademark in India is pending. Our trademarks were obtained between 2006 and 2024. Nearly all of our United States trademarks are federal trademarks.
We operate in an industry characterized by extensive patent litigation. Patent litigation may result in significant damage awards and injunctions that could prevent the manufacture and sale of affected products or result in significant royalty payments in order to continue selling the products.
We undertake reasonable measures to protect our patent rights, including monitoring the products of our competitors for possible infringement of our patents. Protecting our intellectual property rights is important to us, and we plan to continue to maintain and defend our rights regarding our intellectual property.
License Agreements
4C Medical Technologies
On August 30, 2017, and as further amended, we entered into a supply and license agreement (as amended, the “4C Agreement”) with 4C Medical Technologies, Inc. (“4C”), a medical technology company that develops medical devices for the treatment of cardiovascular valve disease. Under the terms of the 4C Agreement, we supply and sell ADAPT® tissue to 4C, to be used in 4C’s production of medical devices related to mitral valves and tricuspid human heart valves and granted a limited license to our related sterilization methods only in connection with use of ADAPT® tissue by 4C in its production of medical devices.
Sales under the 4C Agreement are made pursuant to individual purchase orders at a price per unit based on anticipated annual volume. There are no minimum purchase commitments under the 4C Agreement.
During the term of the 4C Agreement, our supply of ADAPT® tissue to 4C is exclusive, meaning that we agree not to develop, manufacture, or sell certain ADAPT® tissue-based products in the mitral valve or tricuspid valve field other than for 4C without prior written approval. We have received $10.2 million in proceeds through December 31, 2025 (life to date) under the 4C Agreement relating to the sale and supply of ADAPT® tissue-based products to 4C and granting 4C a worldwide license to use our sterilization method in connection with those supplied ADAPT® tissue-based products.
Pursuant to the 4C Agreement, 4C was granted a limited, revocable and royalty free license to use certain of our trademarks for marketing purposes for 4C’s medical devices that use ADAPT® tissue. On October 14, 2019, in light of the transaction with LeMaitre, we revoked 4C’s license to the CardioCel™ trademark only. We retained our intellectual property rights existing at the time of the 4C Agreement (except for limited licenses granted to 4C in effect during the term of the 4C Agreement), including new intellectual property rights relating to our tissue products developed either solely by us or jointly by us and 4C. The last-to-expire patent related to the intellectual property covered by the 4C Agreement is scheduled to expire in 2032.
The 4C Agreement had an initial term that expired on June 1, 2025, and under its terms would automatically renew for successive one-year periods unless either party provided written notice of non-renewal at least 180 days prior to the applicable renewal date. On November 26, 2025, we notified 4C that we would not renew the 4C Agreement for the next renewal term. We will not incur any early termination penalties in connection with its non-renewal of the 4C Agreement. The termination of this contract does not materially impact our financial results.
Collaborations
v2vmedtech
On April 18, 2023, we purchased 30% of the equity capital stock of v2vmedtech, pursuant to a contribution and stock purchase agreement (the “Stock Purchase Agreement”), and concurrently contributed $0.2 million and entered into a series of agreements (collectively, the “v2v Agreements”) with v2vmedtech. v2vmedtech has a license agreement with Columbia University to develop an innovative heart valve repair device utilizing a transcatheter edge-to-edge repair method for a minimally invasive treatment of mitral and tricuspid valve regurgitation, also known as leaky valve.
Under the terms of the v2v Agreements, we agreed to provide certain development services to v2vmedtech in exchange for equity in v2vmedtech. Pursuant to the v2v Agreements, we provide engineering, clinical, regulatory, marketing, and executive management resources, but excluding medical and chief medical officer services, in connection with v2vmedtech’s development of these valve repair devices. We are responsible for developing products and preparing regulatory filings and all costs and expenses incurred by us directly, related to the development of devices constitute development contributions under the v2v Agreements, for which we are solely responsible. These contributions are to be provided over five stages linked to key development and regulatory requirements for the device for transcatheter edge-to-edge repair of the mitral valve (“TEER Product”).
Stage 1 is the development of a preferred concept for the TEER Product, during which we will provide analytical, engineering and product development services for the TEER product, gather and document preliminary or critical product requirements, create product specifications, design at least one concept to meet that product specification, and provide initial prototypes. During this stage, v2vmedtech will also establish a separate medical advisory board (the “v2v Advisory Board”). Stage 1 concluded with a design review with non-Anteris members of v2vmedtech, prior to proceeding to Stage 2. The R&D contributions (excluding general and administration expenses) paid by us under Stage 1 were $2.2 million.
Stage 2 involved manufacturing and testing prototypes of the preferred concept to finalize the TEER Product design for concept lock. This stage included additional engineering and product development services to modify the preferred concept of the TEER Product at our sole discretion. Before we make a decision to advance to Stage 3, a design review with non-Anteris members of v2vmedtech will be conducted and their feedback will be considered. In addition, to advance to Stage 3, the TEER Product must meet all established criteria in our quality system. The R&D contributions (excluding general and administration expenses) paid by us as set out in the Development Agreement under Stage 2 are expected to be $0.4 million to $0.8 million.
Stage 3 involves non-clinical bench lab testing of the TEER Product, at our discretion. Before we make a decision to advance to Stage 4, a design review with non-Anteris members of v2vmedtech will be conducted and their feedback will be considered. The R&D contributions (excluding general and administration expenses) paid by us as set out in the Development Agreement under Stage 3 are expected to be $0.8 million to $1.8 million.
Stage 4 involves preclinical acute and chronic studies of the TEER Product in animals to support regulatory submissions, which will be undertaken at our discretion. Before we make a decision to advance to Stage 5, a design review with non-Anteris members of v2vmedtech will be conducted and their feedback will be considered. Approval from v2vmedtech’s Advisory Board may be required before proceeding to Stage 5. The R&D contributions (excluding general and administration expenses) paid by us as set out in the Development Agreement under Stage 4 are expected to be $0.7 million to $1.6 million.
Stage 5 is the first use of the TEER Product in a first-in-human study in one cohort of patients anywhere in the world. During this stage, v2vmedtech will enter into agreements with the sites and practitioners performing the first-in-human study services and must maintain appropriate insurance. A review of endpoints and resulting data from the first-in-human study will be conducted by us and by appropriate non-Anteris members of v2vmedtech in order to determine the success of the first-in-human study. The R&D contributions (excluding general and administration expenses) paid by us under Stage 5 as set out in the Development Agreement are expected to be $1.0 million to $2.2 million.
During Stages 2 through 5, we may solicit input from the v2v Advisory Board and will coordinate, facilitate and participate in meetings of the v2v Advisory Board. We are generally permitted to use our own employees, resources, lab facilities and other internal resources during the five development stages.
We have an option to terminate our activities for v2vmedtech, subject to certain break rights. These break rights allow us to discontinue additional development contributions subject to a fee of $0.2 million during Stage 1 and incrementally increasing by $0.2 million for each stage of development to a maximum $1.0 million break fee in Stage 5. We will also pay all customary corporate, operational, and legal costs (“operational contributions”) of v2vmedtech up to an annual amount determined by vmedtech’s board of directors. After the earlier of the completion of Stage 5 or the incurrence of $10.0 million of development contributions and operational contributions, our ownership stake in v2vmedtech will be increased from 30% to between 58% and 60%.
v2vmedtech owns all intellectual property rights to the technology and data developed (the “Developed Technology and Data”) pursuant to the v2v Agreements. However, under the terms of the v2v Agreements, v2vmedtech grants us a perpetual and exclusive license to the Developed Technology and Data for medical device applications other than leaky valve devices. As v2vmedtech is a development company, there is no revenue currently generated by this entity.
The v2v Agreements will expire one year after completion of Stage 5. We may terminate the v2v Agreements upon exercise of our break rights under the Stock Purchase Agreement and payment of the applicable break fee or upon a material breach by v2vmedtech. v2vmedtech may terminate the v2v Agreements once we no longer own any shares of v2vmedtech’s issued and outstanding capital stock or upon its exercise of its break rights under the Stock Purchase Agreement or the exercise of certain rights it holds under the Stock Purchase Agreement. We and v2vmedtech may terminate the v2v Agreements upon an event of insolvency or a material breach by the other party.
Development is currently in Stage 2 and has reached concept lock on the clips and coupler. Timing for a FIH trial cannot be reasonably determined at this time as it is contingent on successful completion of further stages of R&D, including the design, prototyping and testing, preclinical testing and completion of regulatory submissions. The timing to complete these activities is influenced by the v2v Agreements, which state that the development agreement can be terminated if certain expenditure amounts, development milestones or regulatory approvals are not incurred or achieved from March 31, 2027 and onwards. The total amount of eligible development contributions and operational contributions paid by us under the v2v Agreements as of December 31, 2025 was $6.2 million.
Single Source Suppliers
Aran Biomedical
We are party to a supply and quality agreement (the “Aran Supply Agreement”), dated November 16, 2021, with Aran Biomedical Teoranta (“Aran”) (subsequently acquired by Integer Holdings Corporation) pursuant to which Aran supplies us with certain knitted materials from time to time pursuant to one or more purchase orders and in accordance with reasonable quality requirements provided by us. The Aran Supply Agreement has an initial term of five years and renews thereafter for successive one-year terms upon mutual written agreement of the parties. Either us or Aran may terminate the Aran Supply Agreement upon an uncured material breach.
Harvey Industries Group
We have entered into a supply and quality agreement (the “Harvey Supply Agreement”) with Harvey Industries Group Pty Ltd (“Harvey”), a supplier of animal derived materials for therapeutic applications. Under the Harvey Supply Agreement, Harvey supplies us with bovine pericardia used in the manufacturing of our products pursuant to orders placed by us. We have the ability to reject any product that does not meet the applicable specifications. The Harvey Supply Agreement expires in May 2026 but may be extended by mutual agreement between us and Harvey. If the Harvey Supply Agreement is not extended, Harvey will continue to supply us with bovine pericardia for an additional four months after the expiration of the Harvey Supply Agreement upon our request. We may terminate the Harvey Supply Agreement without cause upon 90 days written notice, and Harvey may terminate the Harvey Supply Agreement with 12 months written notice. Either us or Harvey may terminate the Harvey Supply Agreement for cause upon an uncured breach or a non-remediable breach.
NPX Medical
We are party to a services agreement (the “NPX Services Agreement”), dated March 25, 2020, and subsequently amended on February 21, 2021 and March 24, 2024, with NPX Medical, LLC (“NPX”), pursuant to which NPX provides certain engineering and manufacturing services to us as requested by us in purchase orders from time to time. NPX also provides certain product development services to us under the NPX Services Agreement. The NPX Services Agreement had an original expiration date of March 25, 2021 and renews automatically for successive one-year terms unless terminated. Either party to the NPX Services Agreement may terminate the agreement without cause upon 30 days written notice to the other party or for cause upon an uncured material breach of the NPX Services Agreement.
We are also party to a quality agreement with NPX (the “NPX Quality Agreement”), dated February 11, 2021, which provides for certain quality requirements for the products manufactured for us by NPX, as specified by us in purchase orders made under the NPX Services Agreement. The NPX Quality Agreement will remain in effect as long as the NPX Services Agreement is in effect.
Switchback Medical
On July 28, 2025, we entered into the First Amended and Restated Master Services Agreement (the “A&R MSA”) with Switchback Medical, LLC (“Switchback”), pursuant to which Switchback provides various development and manufacturing services, including engineering and testing services, pursuant to purchase orders made by us from time to time, at set prices per unit, and in compliance with various quality management and regulatory requirements.
Under the A&R MSA, we granted Switchback a limited, exclusive, revocable, non-sublicensable, fully paid-up, royalty-free license to certain of our intellectual property to be used solely for the purpose of manufacturing products during the term of the A&R MSA. We retain all rights, title and interest in the results of any testing services, reports or data generated or provided by Switchback and to any developed intellectual property.
The A&R MSA expires on March 31, 2028, and will automatically renew for successive one-year terms unless terminated by either the party at least 180 days prior to the end of the then-current renewal term.
Taurus Engineering and Manufacturing
We are party to a supplier quality agreement (the “Taurus Supplier Agreement”), dated February 15, 2024, with Taurus Engineering and Manufacturing, Inc. (“Taurus”), under which Taurus provides us with certain manufacturing services and supplies us with raw materials in accordance with specified quality requirements and other specifications. Taurus is not an exclusive supplier to us for the materials that it supplies, but under the terms of the Taurus Supplier Agreement, Taurus may not supply anyone other than us with the materials covered by the Taurus Supplier Agreement. The Taurus Supplier Quality Agreement had an initial two‑year
term that ended on February 15, 2026. Arrangements are ongoing to extend
the agreement in relation to quality and supply.
Other Agreements
CRF
We are party to a Combined Bioinformatics Master Services Agreement, dated September 1, 2021, with CRF (the “CRF MSA”). Pursuant to the CRF MSA, CRF is engaged on a per project basis to perform independent analyses and provide interpretations on various types of medical data and information, provide comprehensive data coordination and analysis center (“DCAC”) services, manage clinical events and data monitoring committees, and health economics and outcomes research (“HEOR”). Data and other research and results generated or produced by CRF concerning core lab and HEOR activities pursuant to the CRF MSA is jointly owned by us and CRF. The data and other research and results generated or produced by CRF concerning DCAC activities pursuant to the CRF MSA is owned by us. Payment terms under the CRF MSA are set forth in work orders for discrete tasks. The original term of the CRF MSA was through December 31, 2022, and has automatically renewed for subsequent annual terms, with the current term expiring on December 31, 2026. Either party to the CRF MSA may provide notice of termination of the CRF MSA for the subsequent annual period or upon 60 days’ notice.
QMED
We have agreed to be bound by General Terms and Conditions with QMED Consulting A/S (“QMED”), pursuant to which QMED provides certain services to us in accordance with individual service agreements (the “Service Agreements”). Pursuant to the Service Agreements first entered into on July 8, 2024, QMED has agreed to provide us with clinical trial submission support for the EU, including the provision of life science services in the areas of regulatory affairs, training, quality assurance and control, clinical trial consultancy and legal representation. Payment terms and term lengths for discrete tasks and services are set forth in individual Service Agreements. Under the General Terms and Conditions, we may terminate the Service Agreements at our discretion by providing 30 days’ notice, or upon ten days’ notice and payment of a 15% termination fee. Either we or QMED may terminate the Service Agreements upon default or an uncured material breach.
Bright Research
We are party to a Master Services Agreement, dated January 12, 2026 (the “Bright MSA”), with Bright Research Partners, Inc. (“Bright Research”), pursuant to which Bright Research provides services to us in support of various clinical activities identified in applicable statements of work (each, a “SOW”). The Bright MSA has an initial term of five years, however, if any SOWs remain in effect at the end of the term, the agreement will automatically be extended for an additional one-year period, unless earlier terminated. Either party may terminate the Bright MSA, for any reason or for no reason, upon ninety (90) days’ written notice to the other party. The Bright MSA may also be terminated upon an uncured material breach or upon the occurrence of certain insolvency‑related events affecting a party. If a SOW with total fees greater than $1 million is subject to an early termination by us without cause, by Bright Research for cause, or as a result of a change of control, we are required to pay Bright Research an amount equal to 20% of the estimated remaining SOW budget.
Government Regulation
United States FDA Regulation of Medical Devices
Our products are regulated as medical devices in the United States. Accordingly, our products and operations are subject to extensive and ongoing regulation by the FDA under the Federal Food, Drug, and Cosmetic Act (“FDCA”), as well as under other federal, state and local regulatory authorities in the United States. For devices intended for commercial distribution in the United States, the FDA regulates product design and development, preclinical and clinical testing, manufacturing, packaging, labeling, storage, record keeping and reporting, clearance or approval, marketing, distribution, promotion, import and export, and post-marketing surveillance to assure their safety and effectiveness for their intended uses.
Unless an exemption applies, each new medical device we seek to commercially distribute in the United States will require either a premarket notification to the FDA requesting a Section 510(k) clearance, de novo classification, or premarket approval application (“PMA”). Additionally, each significant modification to a 510(k)-cleared or de novo classified device will require a new submission prior to marketing, and each modification that affects the safety and effectiveness of a device with an approved PMA will require a new PMA or supplement. The 510(k) clearance, de novo classification and premarket approval processes can be resource intensive, expensive, and lengthy, and require payment of significant user fees unless a waiver or exemption is available.
FDA classifies medical devices into one of three classes - Class I, Class II or Class III - depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurances with respect to safety and effectiveness.
Class I devices are those for which safety and effectiveness can be reasonably assured by adherence to the FDA’s general controls for medical devices, which include compliance with the applicable portions of FDA’s current good manufacturing practices for devices, establishment registration and device listing, reporting of adverse events and malfunctions, reporting of corrections and removals, and appropriate, truthful and non-misleading labeling and promotional materials. Some Class I devices, called Class I reserved devices, also require premarket clearance by the FDA through the 510(k) premarket notification process described below. Most Class I devices are exempt from the premarket notification requirements. A class I device is not exempt from 510(k) notification requirements if it is intended for a use of substantial importance in preventing impairment of health, or presents a potential unreasonable risk of illness or injury.
Class II devices are those that are subject to the FDA’s general controls and any other special controls deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, patient registries, product-specific FDA guidance documents, special labeling requirements and post-market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA through the 510(k) premarket notification process, although some Class II devices are exempt from such requirement.
Under the 510(k) premarket notification process, a medical device manufacturer provides the FDA with a premarket notification that it intends to begin commercializing a product and demonstrates to the FDA that the product is substantially equivalent to another legally marketed predicate device. To be found substantially equivalent to a predicate device, the device must be for the same intended use and have either the same technological characteristics as the predicate or different technological characteristics that do not raise different questions of safety or effectiveness. In some cases, the submission must include data from clinical studies in order to demonstrate substantial equivalence to a predicate device. Commercialization may commence when the FDA issues a clearance letter finding such substantial equivalence.
Class III devices include those devices that (i) cannot be classified into Class I or Class II because insufficient information exists to determine that general and special controls would provide a reasonable assurance of safety and effectiveness, and (ii) are intended for uses that are life-supporting, life-sustaining, of substantial importance in preventing impairment in human health, or present a potential unreasonable risk of illness or injury.
Additionally, novel devices that lack a predicate device to which they can demonstrate substantial equivalence via the 510(k) premarket notification process are automatically classified into Class III, unless the manufacturer can demonstrate that the device should be classified into Class I or II via the de novo classification process, discussed below. Devices placed in Class III are subject to premarket approval, which requires submission of valid scientific evidence demonstrating a reasonable assurance of the safety and effectiveness of the device for its intended use. The premarket approval process is generally more costly and time consuming than the 510(k) premarket notification process or the de novo classification process. A PMA typically includes, but is not limited to, extensive technical information regarding device design and development, preclinical and clinical trial data, manufacturing information, labeling, and financial disclosure information for the clinical investigators in device studies.
CardioCel™, VascuCel™ and ADAPT® are pericardial tissue products and are Class II medical devices.
CardioCel™ was cleared for marketing by the FDA on January 30, 2014 as a Class II device. A modified version of CardioCel™ was cleared for marketing by the FDA on April 28, 2017. VascuCel™ (another modified version of CardioCel™) was cleared for marketing by the FDA on October 14, 2016. ADAPT® tissue was cleared for marketing by the FDA on April 3, 2020.
Replacement heart valves, including the DurAVR® THV, are Class III medical devices. Additionally, because the ComASUR® Delivery System is required for use of the DurAVR® THV, the ComASUR® Delivery System will be regulated as a component of the DurAVR® THV Class III device (as part of the overall system). Accordingly, the ComASUR® Delivery System will be reviewed under any PMA submitted for the DurAVR® THV System.
As noted above, if a novel device lacks a predicate device to which it can demonstrate substantial equivalence via that 510(k) process, it is automatically classified into Class III, which means it requires a PMA. However, under the de novo classification process, a manufacturer that believes its novel device is actually low to moderate risk, can request the classification of the novel device into Class I or Class II. To obtain de novo classification, the manufacturer must demonstrate that when general controls, or general controls and special controls, are applied, the probable benefits to health from using the device outweigh probable risks of such use, and that a significant portion of the target population will have clinically significant results from use of the device. If a device is de novo classified into Class I or Class II, it becomes a legally marketed predicate device to which future devices can claim substantial equivalence by submitting a 510(k). The de novo classification process is generally more costly and time consuming than the 510(k) premarket notification process but can be less costly and time consuming than the premarket approval process. A de novo classification request typically includes information similar to that required in a PMA, plus a recommendation for the proposed classification (Class I or Class II) and, if the device is proposed to be classified into Class II, any proposed special controls.
Obtaining FDA marketing clearance or approval for medical devices is expensive and uncertain, and may take several years, and generally requires significant scientific and clinical data. Our DurAVR® THV System is classified as a Class III device for which we expect to submit a PMA upon completion of the currently contemplated pivotal clinical trial.
IDE Process
In the United States, absent certain limited exceptions, human clinical trials intended to support medical device clearance or approval require IDE approval. An IDE authorizes distribution of devices that lack premarket approval, de novo classification or 510(k) clearance for clinical evaluation purposes. Some types of studies deemed to present “non-significant risk” are deemed to have an approved IDE if certain requirements are satisfied, including but not limited to obtaining Institutional Review Board (“IRB”) approval for the study before initiation of the study, obtaining informed consent from study subjects, and complying with certain recordkeeping and reporting requirements. If the device presents a “significant risk” to human health, as defined by the FDA, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, such as animal, biocompatibility and laboratory testing results, showing that it is safe to test the device in humans and that the clinical test protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of test subjects. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA, the study protocol and informed consent documents are approved by appropriate IRBs at the clinical trial sites, and informed consent from study subjects has been obtained. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials, and although the FDA’s approval of an IDE allows clinical testing to go forward for a specified number of subjects, it does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and effectiveness, even if the trial meets its intended success criteria.
All non-exempt clinical trials must be conducted in accordance with the FDA’s IDE regulations that govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials must further comply with the FDA’s regulations for IRB approval and for informed consent and other human subject protections. Required records and reports are subject to inspection by the FDA and other applicable authorities.
The results of clinical trials may be unfavorable, or, even if the intended safety and effectiveness success criteria are achieved, may not be considered sufficient for the FDA to grant marketing approval or clearance of a product. The commencement or completion of any clinical trial may be delayed or halted, or be inadequate to support approval of a PMA, for numerous reasons, including the following:
•
the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold;
•
patients do not enroll in clinical trials at the rate expected;
•
patients do not comply with trial protocols;
•
patient follow-up is not at the rate expected;
•
patients die during a clinical trial, even though their death may not be related to the products that are part of the trial;
•
device malfunctions occur in unexpected ways, with unexpected frequency, or with potential adverse consequences;
•
side effects or device malfunctions of similar products already in the market that change the FDA’s view toward approval of new or similar premarket approvals or clearance of new or similar 510(k)s or de novo classification requests, or result in the imposition of new requirements or testing;
•
IRBs and third-party clinical investigators may delay or reject the trial protocol;
•
third-party clinical investigators decline to participate in a trial or do not perform a trial on the anticipated schedule or consistent with the clinical trial protocol, investigator agreement, investigational plan, good clinical practices, the IDE regulations, or other FDA or IRB requirements;
•
we or third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans, or otherwise fail to comply with the IDE regulations governing responsibilities, records, and reports of sponsors of clinical investigations;
•
third-party clinical investigators have significant financial interests related to us or our study such that the FDA deems the study results unreliable, or we or investigators fail to disclose such interests;
•
regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials;
•
changes in government regulations or administrative actions;
•
the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or effectiveness; or
•
the FDA concludes that our trial design is unreliable or inadequate to demonstrate safety and effectiveness.
The Premarket Approval Process
Following receipt of a PMA, the FDA conducts an administrative review to determine whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If it is sufficiently complete, the FDA will accept the application for filing and begin the substantive review. The FDA, by statute and by regulation, has 180 days to review a filed PMA, although the review of an application more often occurs over a significantly longer period of time. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies within the submission communicated by the FDA. The issuance of a deficiency letter automatically stops the FDA 180-day review clock. The FDA considers a premarket approval or premarket approval supplement to have been voluntarily withdrawn if an applicant fails to respond to an FDA request for information (e.g. major deficiency letter) within a total of 360 days. Before approving or denying a PMA, the FDA may hold an advisory committee meeting to obtain advice related to the safety and effectiveness of the medical devices and provide the FDA with the committee’s recommendation on whether the FDA should approve the submission, approve it with specific conditions, or not approve it. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Prior to approval of a PMA, the FDA may conduct inspections of the clinical trial data and clinical trial sites, as well as inspections of the manufacturing facility and processes for the device. Overall, the FDA review of a PMA generally takes between one and two years but may take significantly longer. The FDA can delay, limit or deny approval of a PMA for many reasons, including:
•
the device may not be shown to be safe or effective to the FDA’s satisfaction;
•
the data from preclinical studies and/or clinical trials may be found unreliable or insufficient to support approval;
•
the manufacturing process or facilities may not meet applicable requirements;
•
the proposed labeling is found to be false or misleading;
•
the device is not shown to conform to a required performance standard; or
•
changes in FDA approval policies or adoption of new regulations may require additional data.
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a premarket approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA’s evaluation of a PMA or manufacturing facilities is not favorable, then the FDA will deny the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. Workforce reductions, agency reorganization, and other changes at the FDA could also impact product approval timelines. The premarket approval process can be expensive, uncertain and lengthy, and a number of devices for which the FDA premarket approval has been sought by other companies have never been approved by the FDA for marketing.
New PMAs or premarket approval supplements generally are required for modifications to an approved device that could affect the safety or effectiveness of an approved device, including modifications to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design of the device that has been approved through the premarket approval process. Premarket approval supplements often require submission of the same type of information as an initial PMA, except that the supplement is limited to information needed to support ay changes from the device covered by the approved PMA and may or may not require as extensive technical or clinical data or the convening of an advisory panel, depending on the nature of the proposed change.
In approving a PMA, as a condition of approval, the FDA may also require some form of post-approval study or post-market surveillance. The applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer-term safety and effectiveness data for the device. The FDA may also require post-market surveillance for certain devices cleared under a 510(k) notification or de novo classification, such as implants or life-supporting or life-sustaining devices used outside a device user facility. The FDA may also approve a PMA with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use. Modifications to the manufacturing process, labeling and design for a device which has received approval through the premarket approval process generally require submission of a new PMA or premarket approval supplement prior to marketing.
Ongoing Regulation by the FDA
Even after the FDA permits a device to be marketed, numerous regulatory requirements apply, including:
•
establishment registration and device listing;
•
the Device cGMP, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, production, control, supplier/contractor selection, complaint handling, documentation, and other quality assurance procedures during the manufacturing process;
•
labeling regulations, advertising and promotion requirements, restrictions on sale, distribution or use of a device, each including the FDA general prohibition against the promotion of products for any uses other than those cleared or approved by the FDA, which are commonly known as “off label” uses;
•
medical device reporting regulations requiring that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or if their device malfunctioned and the device or a similar device marketed by the manufacturer would be likely to cause or contribute to a death or serious injury if the malfunction were to recur;
•
medical device corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections or removals if undertaken to reduce a risk to health posed by a device or to remedy a violation of the FDCA that may present a risk to health;
•
recall requirements, including a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death;
•
any order from FDA to repair, replace or refund a device;
•
product export requirements;
•
device tracking requirements; and
•
post-market study and surveillance requirements.
If a device receives 510(k) clearance or de novo classification, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new 510(k) clearance or possibly a premarket approval. The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with the manufacturer’s determination not to seek a new 510(k) clearance, the FDA may retroactively require the manufacturer to seek 510(k) clearance or possibly a premarket approval. The FDA could also require a manufacturer to cease marketing and distribution and/or to recall the modified device until 510(k) clearance or a premarket approval is obtained. Also, in these circumstances, a manufacturer may be subject to more significant actions, including regulatory fines and penalties.
Some changes to an approved premarket approval device, including changes in indications, labeling, or manufacturing processes or facilities, among others, generally require submission and FDA approval of a new PMA or premarket approval supplement, as appropriate, before the change can be implemented. Supplements to a PMA often require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to that information needed to support the proposed change from the device covered by the original premarket approval. The FDA generally uses the same procedures and actions in reviewing premarket approval supplements as it does in reviewing original PMAs, although some premarket approval supplements may be approved more quickly, such as supplements describing certain modifications in the manufacturing process that do not affect the specifications of the device.
FDA regulations require us to register as a medical device manufacturer with the FDA. These regulations require that we manufacture our products and maintain related documentation in a prescribed manner with respect to manufacturing, testing and control activities. Furthermore, the FDA requires us to comply with various FDA regulations regarding labeling. Our facilities, records and manufacturing processes are subject to periodic unscheduled inspections by the FDA or other regulatory authorities. When the FDA conducts an inspection, the investigators will identify any deficiencies they believe exist in the form of a notice of inspectional observations, or Form FDA 483. If we receive a notice of inspectional observations or deficiencies from the FDA following an inspection, we would be required to respond in writing and would be required to undertake corrective and/or preventive or other actions in order to address the FDA’s or other regulators’ concerns. Failure to address the FDA’s concerns may result in the issuance of a warning letter or other enforcement or administrative actions described below.
Additionally, some states have enacted laws and regulations governing the manufacture, sale, marketing or distribution of medical devices. These laws and regulations may also require medical device manufacturers and/or distributors doing business within multiple states to register or apply for state licenses. These laws and regulations could also subject our facility to state inspection on a routine basis for compliance with any applicable state requirements.
Failure by us or by our suppliers to comply with applicable federal or state regulatory requirements can result in enforcement action by the FDA or state authorities, which may include any of the following sanctions:
•
warning or untitled letters, fines, injunctions, consent decrees and civil penalties;
•
unanticipated expenditures, including for repairs, replacements, or refunds of devices;
•
customer notifications, voluntary or mandatory recall or seizure of our products;
•
operating restrictions, partial suspension or total shutdown of production;
•
delay in reviewing, or refusal to clear or approve, submissions or applications for new products or modifications to existing products;
•
FDA refusal to issue certificates to foreign governments needed to export products for sale in other countries;
•
suspension or withdrawal of FDA approvals or clearances that have already been granted; and
•
criminal prosecution.
Newly discovered or developed safety or effectiveness data may require changes to a product’s labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation or regulations, may be established, or the FDA’s policies may change, which could delay or prevent regulatory clearance or approval of our products under development.
Regulation of Medical Devices Outside the United States
Outside of the United States, the regulation of medical devices is also complex. In Europe, for instance, products are subject to extensive regulatory requirements. In 2021, a new regulatory scheme for medical devices, namely the EUMDR, became effective in EU Member States subject to a transition period during which some devices that were in conformity with the previous rules could still be placed on the EU market for some time. The EUMDR requires that medical devices may only be placed on the market or put into service if they meet certain pre-established general safety and performance requirements when properly installed, maintained, and used in accordance with their intended purpose. The EUMDR has significant requirements for many medical devices, including requirements for clinical evidence necessary to demonstrate the devices’ conformity (and the related documentation), device identification and traceability, registration of devices and of economic operators throughout the distribution chain and post-market surveillance (dealing with the collection and review of the experience gained from devices for the purpose of identifying any for any necessary corrective or preventive actions after they have been placed on the market or put into service). In some regions, the level of government regulation of medical devices is increasing, which can lengthen time to market and increase registration and approval costs. In many countries, the national health or social security organizations require products to be qualified before they can be marketed and considered eligible for reimbursement.
In many instances, global regulatory agencies have come together in an attempt to harmonize medical device regulatory requirements. In 2011, the regulatory agencies of Australia, Brazil, Canada, China, European Union, Japan and the United States, as well as the World Health Organization came together and established the International Medical Device Regulators Forum (the “IMDRF”). The IMDRF continues to grow and now has a management committee of regulatory agency representatives from 11 countries as well as the European Union and affiliate members and observers from many other countries. One example of the IMDRF harmonizing medical device regulatory requirements is the Medical Device Single Audit Program, whereby a medical device manufacturer can have a single Quality Management System audit of their facility which covers the regulatory requirements of Australia, Brazil, Canada, Japan and the United States. Instead of having separate periodic quality inspections from regulators of each of these countries, a single comprehensive inspection is performed.
Other regional groups working to harmonize regulatory requirements are the Asia-Pacific Economic Cooperation group, Global Harmonization Working Party and African Medical Devices Forum. While regulatory requirements are constantly evolving, regulatory agencies recognize the impact and are attempting to harmonize their efforts.
While the list of regulated countries continues to grow, many of the regulated countries leverage device approvals from the United States or Europe, meaning that the testing and clinical studies required to satisfy device safety and efficacy requirements of the United States and Europe, often carry over to other geographies.
Other United States Regulatory Matters
Medical device companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Manufacturing, sales, promotion, third-party payor reimbursement and other activities following product clearance or approval are subject to regulation by numerous regulatory authorities in the United States in addition to the FDA, including the Centers for Medicare and Medicaid Services (“CMS”), other divisions of the Department of Health and Human Services (“HHS”), the Department of Justice, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency, and state and local governments. For example, in the United States, sales, marketing, participation in government health care programs or contracts with third-party payors, and scientific and educational programs also must comply with state and federal fraud and abuse, anti-kickback, false claims, transparency, government price reporting, anti-corruption, and health information privacy and security laws and regulations. Internationally, other governments also impose regulations in connection with their healthcare reimbursement programs and the delivery of healthcare items and services. These laws include the following:
•
United States federal healthcare fraud and abuse laws generally apply to activities involving products and services that are covered under federal healthcare programs such as Medicare and Medicaid. The federal Anti-Kickback Statute (the “Anti-Kickback Statute”) makes it a crime for any person, including a prescription medical device manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce or reward referrals, including the purchase, recommendation, order or prescription of a particular medical device, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Almost any financial arrangement with a healthcare provider, patient or customer could implicate the Anti-Kickback Statute. Statutory exceptions and regulatory safe harbors protect certain arrangements if specific requirements are met. Individual states have corollaries to the federal Anti-Kickback Statute that may also apply and may be more expansive or impose additional requirements.
•
Another fraud and abuse law that may be implicated by ownership and compensation arrangements with physicians or their families is the Physician Self-Referral Law, commonly referred to as the “Stark Law.” The Stark Law prohibits physicians from referring patients to receive “designated health services” payable by Medicare or Medicaid from entities with which the physician or an immediate family member has a financial relationship, unless an exception applies. Individual states have corollaries to the federal Stark law that may also apply and may be more expansive or impose additional requirements.
•
Another development affecting the medical technology industry is the increased use of the federal Civil False Claims Act (the “False Claims Act”) and, in particular, actions brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. In recent years, the number of suits brought against healthcare companies by private individuals has increased dramatically. The federal civil and criminal false claims acts prohibit individuals or entities from knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. Individual states have false claims acts with respect to Medicaid spending that may also apply and may be more expansive or impose additional requirements. Additionally, some states have fraud provisions that apply to commercial payors or all payors under insurance laws that have similar whistleblower or relator provisions (e.g., Insurance Fraud Prevention Act for California).
•
The federal Civil Monetary Penalty Law (“CMP”) allows the HHS Office of Inspector General to seek civil monetary penalties and sometimes exclusion from participation in the government health care programs for a wide variety of conduct. For example, the CMP and implementing regulations impose penalties against any person or entity that is determined to have presented or caused to be presented a claim to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Other conduct that may result in violation of the CMP is offering or transferring remuneration to a federal healthcare program beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier.
•
The Health Insurance Portability and Accountability Act (“HIPAA”) prohibits executing or attempting to execute a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”) and their implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information. While HIPAA applies only to covered entities, which generally does not include device manufacturers, HIPAA and HITECH impose those same obligations to business associates under contractual terms. HIPAA may also still apply directly to the manufacturer depending on the scope and nature of data sharing arrangement or other contracting arrangements. In addition to HIPAA and its accompanying regulations, device manufacturers may be subject to additional state consumer and privacy laws which may be more expansive or restrictive on the use and protection of patient and consumer data.
•
The FDCA prohibits the adulteration or misbranding of medical devices. Medical device manufacturers may also be subject to state corollaries to the FDCA.
•
The federal Physician Payment Sunshine Act and its implementing regulations, which require applicable manufacturers of covered drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS and HHS information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), non-physician healthcare professionals (such as physician assistants and nurse practitioners, among others) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members.
•
The Foreign Corrupt Practices Act (“FCPA”) prohibits any United States individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring us to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, if any, and to devise and maintain an adequate system of internal accounting controls for international operations. On February 10, 2025, Executive Order “Pausing Foreign Corrupt Practices Act Enforcement to Further American Economic and National Security” was signed, which directs the U.S. attorney general to review and update guidelines and policies related to FCPA enforcement and to cease new FCPA investigations and enforcement actions until a new enforcement policy is implemented.
•
Analogous state and foreign laws and regulations, such as state anti-kickback, anti-referral, and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers or paid for by patients directly; state laws that require certain medical device companies to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require applicable manufacturers to disclose or report certain information related to payments and other transfers of value to health care professionals and entities or sales, marketing, pricing, clinical trials, marketing expenditures and activities, and state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts; and state laws related to insurance fraud in the case of claims involving private insurers.
United States Health Care Reform
Changes in healthcare policy in the United States subject us to additional regulatory requirements that may interrupt the development and the commercialization of our current and future products. Current and future legislative proposals may limit coverage for the procedures associated with the use of our products. Changes in healthcare policy may further reform state and federal legislation and regulations related to reimbursement and coverage for our current and future products.
We believe that there will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to disclose and/or reduce health care costs while expanding individual healthcare benefits. Certain of these changes could impose additional limitations on the rates we will be able to charge for our current and future products or the amounts of reimbursement available for our current and future products from governmental agencies or third-party payors. Federal and state regulators are also prioritizing costs and charge transparency initiatives, including rebate programs, that may impact our ability to charge and collect payment for our products or charging and collection activities for services that use our products. Other initiatives currently on the healthcare reform agenda include value-based care initiatives, which will impact medical device sales and contracting models, and therefore, product pricing. Such health policy priorities are consistently evolving and subject to change under shifting political conditions and leadership. As such, depending on policy priorities, current and future health care reform legislation and policies could have a material adverse effect on our business and financial condition given the potential impact to the availability and demand for our products. Notably, we will be impacted by the reimbursement coverage eligibility and rate schedules set by CMS for both our products and for services and procedures involving our products. For example, on June 21, 2019, CMS issued a National Coverage Determination for Transcatheter Aortic Valve Replacement which informed Medicare Administrative Contractors of coverage requirements for the procedure. Current coverage and reimbursement levels are subject to ongoing analysis and could change, thus having an adverse effect on market demand and our pricing flexibility.
Data Privacy and Security
Numerous state, federal and foreign laws govern health privacy, consumer protection, and other use of individually identifiable information. This includes the collection, dissemination, use, access to, confidentiality and security of personal information and health-related information. In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws, including HIPAA, and federal and state consumer protection laws and regulations, that govern the collection, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our partners. Notably, the Office for Civil Rights at HHS has expanded the application of HIPAA to regulated entities’ use of tracking technologies that collect and analyze information about how users interact with regulated entities’ websites or mobile applications. In addition, certain state and non-United States laws and regulations, such as the California Consumer Privacy Act, the California Privacy Rights Act and the EU General Data Protection Regulation, govern the privacy and security of personal information, including health-related information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws or regulations, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
Corporate Information
The Company was incorporated in the State of Delaware on January 29, 2024. The Company is a global company with its principal executive offices located at Toowong Tower, Level 3, Suite 302, 9 Sherwood Road, Toowong, QLD 4066, Australia, and other key locations located at 860 Blue Gentian Road, Suite 340, Eagan, Minnesota 55121 as well as two other sites in Minnesota and sites in Western Australia, Australia and Geneva, Switzerland. The Company’s telephone number is +61 7 3152 3200. Additional information can be found on our website address: www.anteristech.com. Information contained on, or that is accessible through, the website shall not be deemed incorporated into and is not a part of this Form 10-K.
Initial Public Offering and Reorganization
On December 12, 2024, we completed our initial public offering pursuant to which we issued and sold 14,878,481 shares of Common Stock at a public offering price of $6.00 per share. We received net proceeds of $80.0 million, after deducting the underwriting discounts, commissions and offering expenses and giving effect to the exercise of the underwriters’ option to purchase additional shares.
Prior to the consummation of the initial public offering, we completed the Reorganization pursuant to which we received all of the issued and outstanding shares of ATPL, which was formerly an Australian public company originally registered in Western Australia, Australia and listed on the Australian Securities Exchange (“ASX”), pursuant to a scheme of arrangement under Australian law between ATPL and its shareholders (the “Scheme”) under Part 5.1 of the Australian Corporations Act 2001 (Cth) (the “Corporations Act”). Contemporaneously with implementation of the Scheme, ATPL also cancelled all existing options it had on issue in exchange for our company issuing replacement options to acquire Common Stock pursuant to a scheme of arrangement between ATPL and its optionholders (the “Option Scheme”) under Part 5.1 of the Corporations Act. The Scheme was approved by ATPL’s shareholders at a general meeting of shareholders, which was held on December 3, 2024. The Option Scheme was approved by ATPL’s optionholders at a general meeting of optionholders held on the same day. ATPL obtained approval of the Scheme and the Option Scheme by the Supreme Court of Queensland on December 4, 2024. As a result of the Reorganization, ATPL became a wholly owned subsidiary of our company and the shareholders of ATPL immediately prior to the consummation of the initial public offering, became holders of either one share of Common Stock or one CDI for every ordinary share of ATPL held as of the record date fixed for the relevant meeting.
2025 Private Placement
On or about October 23, 2025 we entered into (i) subscription agreements (the “Subscription Agreements”) with certain investors, pursuant to which we issued and sold 2,346,936 shares (the “PIPE Shares”) of Common Stock, each with an accompanying warrant (the “Common Stock Warrants”) to purchase one share of Common Stock, at a price of $4.90 per share of Common Stock and accompanying Common Stock Warrant (the “Common Stock Offering”), and (ii) confirmation letters (the “Confirmation Letters”) with certain investors, pursuant to which we issued and sold 2,788,064 CDIs, each with an accompanying warrant (the “CDI Warrants”) to purchase one CDI, at a price of A$7.50 per CDI and accompanying CDI Warrant (the “CDI Offering”, and together with the Common Stock Offering, the “2025 Private Placement”). The 2025 Private Placement generated gross proceeds totaling approximately $25.2 million. The Common Stock Offering closed on October 27, 2025 and the CDI Offering closed on November 5, 2025. We also issued 250,000 CDI Warrants to the lead manager of the 2025 Private Placement.
The issuance and sale of the PIPE Shares, Common Stock Warrants, CDIs and CDI Warrants pursuant to the Subscription Agreements and Confirmation Letters were not, and the issuance of the Common Stock Warrant Shares and CDI Warrant Shares will not be, registered under the Securities Act and were and will be issued and sold in reliance on the exemption provided by Section 4(a)(2) of the Securities Act, including under Rule 506 of Regulation D promulgated thereunder, with respect to the PIPE Shares, the Common Stock Warrants and the Common Stock Warrant Shares, and Regulation S with respect to the CDIs, CDI Shares, CDI Warrants, and CDI Warrant Shares.
Each of the Common Stock Warrants and the CDI Warrants are exercisable commencing six months following the date of issuance and expires five years from issuance.
2026 Public Offering
On January 22, 2026, we completed an underwritten public offering (the “2026 Public Offering”) of 40,000,000 shares of our Common Stock, which included the full exercise of the underwriters’ option to purchase additional shares, at a public offering price of $5.75 per share. The 2026 Public Offering generated gross proceeds of approximately $230.0 million, prior to deducting underwriting discounts and commissions and estimated offering expenses.
The 2026 Public Offering was made pursuant to our shelf registration statement on Form S-3 (Registration No. 333-292565), which was previously filed with the SEC and declared effective on January 8, 2026, and a prospectus supplement dated January 20, 2026.
Medtronic Private Placement
On January 20, 2026, we entered into a stock purchase agreement with Covidien Group S.à r.l. (“Covidien”), a wholly owned subsidiary of Medtronic plc (together with Covidien, “Medtronic”), pursuant to which we issued and sold to Medtronic 15,652,173 shares of Common Stock at a purchase price of $5.75 per share (the “Medtronic Private Placement”). The Medtronic Private Placement closed on January 22, 2026, immediately after the completion of the 2026 Public Offering, and generated gross proceeds of approximately $90.0 million, before deducting placement agent fees and estimated offering expenses.
The issuance and sale of the shares of Common Stock to Medtronic in the Medtronic Private Placement was not registered under the Securities Act and were issued and sold in reliance on the exemption provided by Section 4(a)(2) of the Securities Act.
Registration Rights Agreement
In connection with the Medtronic Private Placement, we entered into a registration rights agreement, dated January 22, 2026, with Medtronic (the “Registration Rights Agreement”), pursuant to which we agreed to file a registration statement covering the resale of the Common Stock sold to Medtronic in the Medtronic Private Placement by Medtronic no later than 18 months following the closing of the Medtronic Private Placement. In addition, pursuant to the Registration Rights Agreement, the Company granted Medtronic the right to demand the sale of the Common Stock sold in the Medtronic Private Placement in one underwritten offering. The Company also agreed to be responsible for all fees and expenses incurred in connection with such registration. See “Item 13. Certain Relationships and Related Transactions, and Director Independence” of Part III of this Form 10-K for further details of the Registration Rights Agreement.
Investor Rights Agreement
Also in connection with the Medtronic Private Placement, we entered into an investor rights agreement, dated January 22, 2026, with Medtronic (the “Investor Rights Agreement”), pursuant to which we and Medtronic have certain rights and obligations, including: (i) Medtronic’s participation rights with respect to future issuances of our equity securities, (ii) transfer restrictions on the shares of Common Stock purchased by Medtronic, (iii) collaboration rights between us and Medtronic, (iv) customary standstill provisions, (v) Medtronic’s right to designate one non-voting board observer to our Board, and (vi) Medtronic’s right to negotiate should we receive certain acquisition proposals. See “Item 13. Certain Relationships and Related Transactions, and Director Independence” of Part III of this Form 10-K for further details of the Investor Rights Agreement.
Human Capital
Overview
As of December 31, 2025, we had approximately 174 full time equivalent employees. We have never experienced a work stoppage or interruption due to labor disputes. We believe our relations with our employees are good.
Employee Talent and Retention
Our business and future operating results depend in significant part upon the continued contributions of our key personnel, including qualified personnel with medical device and tissue processing experience, and senior management with experience in the medical device or tissue processing space, many of whom would be difficult to replace. Our business and future operating results depend in significant part on our ability to attract and retain qualified management, operational, clinical, regulatory, marketing, sales, and support personnel for our operations.
We have programs and processes in place to help ensure that our compensation, benefits programs, and work environment attract and retain such personnel, and we strive to enhance those programs and processes to respond to the increasingly competitive market for talent. We also strive to offer competitive, equitable pay, comprehensive benefits, and services that retain and meet the varying needs of our employees. The principal purposes of our equity and cash incentive plans and non-officer incentive plans are to attract, retain, motivate, and reward our employees.
Culture
Fostering and maintaining a strong and collaborative culture is a key strategic focus. We also have ethics and compliance policies that are designed to instill a commitment to ethical behavior and legal compliance across our company. Employees are encouraged to approach their supervisors if they believe violations of policies have occurred. Employees are also able to confidentially and anonymously report any such violations through ethics and compliance hotlines and an online form. Furthermore, the company has a whistleblower policy whereby employees are able to submit an anonymous disclosure either by email, web form or our ethics and compliance hotlines.
We hire based on our AORTIC (Accountability, Objectivity, Respect, Teamwork, Integrity, Courage) values and continuously build our culture around those values. Additionally, employees have an annual goal focused on demonstrating our AORTIC Values. Employees are encouraged to present culture building activities that promote collaboration and inclusivity. We support employee engagement through a grassroots social committee that promotes AORTIC-based activities and through EmpowHer, an international employee initiative supporting inclusive work cultures. We also maintain CEO-level recognition programs to acknowledge employees who demonstrate a strong commitment to our values.
Training and Development
We consider talent management and development programs instrumental to sustaining a high performing, highly engaged workforce, and we endeavor to make these opportunities accessible to employees at all levels. In 2025 we increased our commitment to employees through investments in developing our people leaders by launching a multi-year new Manager Essentials Program curriculum. The program is focused on building and enhancing communication, engagement, retention and overall people management capabilities. In addition, we offer individual contributor and role specific development programs, including educational workshops and department-led, knowledge-based training (such as quality systems, safety, simulation demonstrations, and participation in professional conferences), to help all employees succeed in their roles and enable their career aspirations.
Health and Safety
We are committed to providing a safe working environment compliant with all relevant and applicable laws. We maintain our commitment to a safe working environment by routinely conducting assessments of the workplace in order to detect, assess and respond to identified hazards or risks; giving preference to removing any hazards or risks in order to prevent injury, illness or incidents from occurring; and striving to reduce the likelihood of the risk or hazard occurring and its severity, where we are unable to eliminate the risk entirely. We have processes to report all work-related injuries, illness and near-misses to management.
Responsibilities of employees and managers are to create and maintain a safe working environment by reporting any unsafe conditions or potential hazards immediately for assessment and remediation; following all safe work method statements, safe travel practices, procedures, instructions, rules legislation and laws relating to workplace health and safety; treating all breaches of workplace health and safety standards seriously and taking appropriate action; and providing adequate information, instruction, training and supervision to enable our employees to perform their roles effectively and safely.
Employee Engagement
Through multiple channels and with the support of a third party provider, we gather anonymous employee feedback to assess satisfaction and engagement and to identify opportunities for improvement. Employee feedback is also gathered through new hire surveys, the employee performance review process, and exit interviews.
Available Information
We file Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, proxy statements, and related amendments, exhibits and other information with the SEC. You may access and read our filings without charge through the SEC’s website at www.sec.gov, the ASX’s website at www.asx.com.au or through our website at https://anteristech.com/investors/financials.html, as soon as reasonably practicable after such materials are electronically filed with or furnished to the SEC pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”) and the ASX Listing Rules. Information contained on, or accessible through, our website shall not be deemed incorporated into and is not a part of this Form 10-K.
Risks Related to Our Business
Risks Related to Our Operating History and Financial Position
We have a history of operating losses and may not achieve or maintain profitability in the future.
We have experienced recurring operating losses and negative cash flows from operating activities since inception. For the years ended December 31, 2025 and 2024, we had total losses after income tax of $94.2 million and $76.0 million, respectively, and negative cash flows from operating activities of $77.8 million and $61.2 million, respectively. As of December 31, 2025 and December 31, 2024, we had an accumulated deficit of $370.5 million and $276.4 million, respectively. We expect to continue to incur additional losses for the foreseeable future. The losses and negative cash flows have primarily been due to the substantial investments we have made to develop our products, costs related to our sales and marketing efforts, costs related to clinical and regulatory initiatives to obtain marketing approval, and infrastructure improvements.
We are a clinical-stage medical device company focused on the development and commercialization of innovative minimally invasive devices to treat heart valve diseases. The success of any product development is uncertain. We expect our operating expenses to increase in the future as we grow our business, including the continuing development and potential future commercialization of DurAVR® THV System, as well as continuing to invest in R&D. Moreover, there is a substantial risk that we may not be able to complete the development of DurAVR® THV System or develop other products. It is possible that none of our products will be successfully commercialized and, if that were to be the case, this would prevent us from ever achieving profitability.
We may also encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors and risks frequently experienced by early-stage medical technology companies in rapidly evolving fields. In addition, as a public company, we incur significant legal, accounting and other expenses. Accordingly, we expect to continue to incur significant operating losses for the foreseeable future and we cannot assure you that we will achieve profitability in the future or that, if we do become profitable, we will sustain profitability. Our failure to achieve and sustain profitability in the future will make it more difficult to finance our capital requirements needed to operate our business and accomplish our strategic objectives, which would have a material adverse effect on our business, financial condition and results of operations and could cause the market price of our Common Stock to decline.
To become and remain profitable, we must succeed in identifying, developing, conducting successful clinical trials for obtaining regulatory clearance and approval for, and eventually commercializing, manufacturing and supplying products, including DurAVR® THV System, that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing clinical trials and preclinical studies of our products, continuing to discover and develop additional products, obtaining regulatory clearance and approval for any products that successfully complete clinical trials, developing manufacturing processes and methods, devising and implementing processes for transferring technology and manufacturing processes to a network of third-party manufacturing sites, establishing necessary quality control, establishing marketing capabilities, and commercializing and ultimately selling any approved products. We may never succeed in any or all of these activities and, even if we do, we may never generate revenue that is sufficient to achieve profitability. Even if we do achieve profitability, we may not be able to sustain profitability or meet outside expectations for our profitability. If we are unable to achieve or sustain profitability or to meet outside expectations for our profitability, the price of our Common Stock could be materially adversely affected.
Because of the numerous risks and uncertainties associated with the development of medical device products, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA or comparable foreign regulatory authorities to perform studies in addition to those we currently anticipate, or if there are any delays in commencing or completing our clinical trials or the development of any of our products, our expenses could increase and commercial revenue could be further delayed and become more uncertain, which will have a material adverse impact on our business.
We may encounter difficulties in managing our growth, which could negatively impact our operations.
We have experienced rapid growth and expect to continue to grow in the future. As we advance our clinical development programs for our products, seek regulatory clearance and approval in the United States and elsewhere and increase the number of ongoing product development programs, we anticipate that we will need to increase our product development, scientific and administrative headcount. Due to the complexity in managing a company that has scaled very quickly and anticipates continued growth, we may not be able to scale our headcount and operations effectively to manage the expansion of our product pipeline or recruit and train the necessary additional personnel. As our operations expand, we also expect that we will need to manage additional relationships with various strategic partners, suppliers and other third parties. We will also need to establish commercial capabilities in order to commercialize any products that may be approved. Such an evolution may impact our strategic focus and our deployment and allocation of resources.
Our ability to manage our operations and growth effectively depends upon the continual improvement of our procedures, reporting systems and operational, financial and management controls. We may not be able to implement administrative and operational improvements in an efficient or timely manner and may discover deficiencies in existing systems and controls. If we do not meet these challenges, we may be unable to execute our business strategies and may be forced to expend more resources than anticipated addressing these issues.
In addition, in order to continue to meet our obligations as a publicly listed company in both Australia and the United States and to support our anticipated long-term growth, we will need to continue to increase our general and administrative capabilities. Our management, personnel and systems may not be adequate to support this future growth.
If we are unable to successfully manage our growth and the increased complexity of our operations, our business, financial position, results of operations and prospects may be harmed.
Unstable market and economic conditions, including as a result of geopolitical events, including current and potential conflicts, may have serious adverse consequences on our business, financial condition, results of operations or liquidity, either directly or through adverse impacts on certain of the third parties on which we rely to conduct certain aspects of our preclinical studies or clinical trials.
Our results of operations could be adversely affected by general conditions in the global economy and in the global financial markets. From time to time, the global credit and financial markets have experienced extreme volatility and disruptions, including severely diminished liquidity and credit availability, rising inflation and monetary supply shifts, rising interest rates, labor shortages, declines in consumer confidence, declines in economic growth, increases in unemployment rates, recession risks, and uncertainty about economic and geopolitical stability. Global economic and business activities face widespread uncertainties. Further, following the transition to a new U.S. presidential administration in January 2025, there have been changes implemented to U.S. tax, fiscal, trade, healthcare, and other government regulatory policy. These and any future policy changes are difficult to predict, could affect the geopolitical landscape, and give rise to circumstances that are outside of our control. There can be no assurance that future deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. A severe or prolonged economic downturn, or additional global financial or political crises, could result in a variety of risks to our business, including delayed clinical trials or preclinical studies, delayed approval of our products, delayed ability to obtain patents and other intellectual property protection, weakened demand for our products, if approved, or our ability to raise additional capital when needed on acceptable terms. If the current equity and credit markets deteriorate, it may make any necessary debt or equity financing more difficult, more costly and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers, suppliers and other partners may not survive an economic downturn, which could directly affect our ability to attain our operating goals on schedule and on budget.
Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business. Furthermore, our stock price may decline due in part to the volatility of the stock market and the general economic downturn.
Additionally, in February 2022, Russia commenced a military invasion of Ukraine. The ongoing conflict and political and physical conditions in Ukraine and Russia, as well as in neighboring countries, may disrupt our EMBARK study in Tbilisi, Georgia, including the ability of third parties on which we rely to perform in accordance with our expectations. Moreover, our ability to conduct 12-month follow-ups with our study participants may be adversely affected as a result of the ongoing conflict, which could significantly delay our clinical development plans and potential clearance or approval of products or cause us to increase our R&D expenses to conduct one or more additional studies, any of which could increase our costs and jeopardize our ability to successfully commercialize our products, if approved.
Risks Related to Our Industry
Unsuccessful clinical trials or procedures relating to products could have a material adverse effect on our future prospects.
The regulatory clearance and approval process for new products and new intended uses for existing products can require extensive clinical trials and feasibility studies. Unfavorable or inconsistent clinical data from current or future clinical trials or procedures conducted by us or third parties, or perceptions regarding this clinical data, could adversely affect our ability to obtain necessary clearances and approvals and the market’s view of our future prospects. Such clinical trials and procedures are inherently uncertain and there can be no assurance that these trials or procedures will be enrolled or completed in a timely or cost-effective manner or result in positive clinical data or a commercially viable product. Clinical trials or procedures may experience significant setbacks even if earlier trials have shown promising results. Furthermore, preliminary results from clinical trials or procedures may be contradicted by subsequent analyses. In addition, results from our clinical trials or procedures may not be supported by actual long-term studies or clinical experience. If preliminary clinical results are later contradicted, or if initial results cannot be supported by actual long-term studies or clinical experience, our business could be adversely affected. Clinical trials or procedures may be delayed, suspended or terminated by us, the FDA or other regulatory authorities at any time if it is believed that the trial participants face unacceptable health risks or any other reasons, and any such delay, suspension, or termination could have a material adverse effect on our prospects or the market’s view of our future prospects.
In particular, our lead product, DurAVR® THV System, is undertaking clinical trials designed to provide the primary clinical evidence on which the FDA could base a decision for premarket approval required for commercialization of the DurAVR® THV System in the United States. There can be no assurance that we will successfully complete the clinical trials and obtain premarket approval for the DurAVR® THV System.
If we are unable to successfully identify, develop, obtain and maintain regulatory clearance or approval for and ultimately commercialize any of our current or future products, or experience significant delays in doing so, our business, financial condition and results of operations will be materially adversely affected.
The clinical development, manufacturing, sales and marketing of our products are subject to extensive regulation as medical devices by regulatory authorities in the United States, the United Kingdom, the European Union, Australia and elsewhere. Our ability to generate revenue from sales of any of our products depends heavily on the successful identification, development, regulatory clearance or approval for and eventual commercialization of any products. All of our products, including the DurAVR® THV, will require significant preclinical and clinical development, regulatory clearance or approval, establishment of sufficient manufacturing supply, including commercial manufacturing supply, and may require us to build a commercial organization and make substantial investment and significant marketing efforts before we generate any revenue from product sales. We are not permitted to market or promote any of our products before we receive regulatory clearance or approval from the FDA or comparable foreign regulatory authorities. Obtaining regulatory clearances and approvals for new products and manufacturing processes can take a number of years and involve expenditure of substantial resources, and, despite the substantial time and expense invested, we may never receive such regulatory clearance or approval for the DurAVR® THV System or other products. The development and commercialization of our products is subject to many risks, including:
•
additional clinical trials may be required beyond what we currently expect;
•
the risk that our financial and other resources are not sufficient to complete the necessary clinical trials;
•
regulatory authorities may disagree with our interpretation of data from our clinical trials or may require that we conduct additional trials;
•
we may be unable to obtain and maintain regulatory clearance or approval of our products in any jurisdiction;
•
regulatory authorities may identify deficiencies in manufacturing processes;
•
regulatory authorities may lack sufficient resources to timely and completely address applications for regulatory clearance or approval of our products;
•
regulatory authorities may change their clearance or approval policies or adopt new regulations;
•
we, or our third-party manufacturers, may not be able to source or produce current Good Manufacturing Practice (“cGMP”) materials for the production of our products or product candidates;
•
our products, if approved, may not be able to be manufactured at a cost or in quantities necessary to make commercially successful products;
•
we may experience delays in the commencement of, enrollment of patients in and timing of our clinical trials or we may not be able to complete our clinical trials;
•
we may not be able to achieve and maintain compliance with all regulatory requirements applicable to our products or operations;
•
we may not be able to maintain a continued acceptable safety profile of our products following clearance or approval;
•
the market may not accept our products, if approved;
•
we may be unable to establish and maintain an effective sales and marketing infrastructure, either through the creation of a commercial infrastructure or through strategic collaborations, and the effectiveness of our own or any future strategic collaborators’ marketing, sales and distribution strategy and operations will affect our profitability;
•
we may experience competition from existing products or new products that may emerge;
•
we may be unable to successfully obtain, maintain, defend and enforce intellectual property rights important to protect our products; and
•
we may not be able to obtain and maintain coverage and adequate reimbursement from third-party payors.
If any of these risks materialize, we could experience significant delays or an inability to successfully develop and commercialize our products, which would have a material adverse effect on our business, financial condition and results of operations.
The successful development of our pipeline of products is highly uncertain and requires significant expenditures and time. In addition, obtaining necessary government clearances and approvals is time-consuming and not assured. If we do not obtain the necessary regulatory clearances or approvals, then we would be unable to commercialize our products.
We currently have a number of products, including the DurAVR® THV System, in development. We conduct extensive preclinical studies and clinical trials to demonstrate the safety and efficacy in humans of our products in order to obtain regulatory clearance or approval for the sale of our products. Preclinical studies and clinical trials are expensive, complex, can take many years and have uncertain outcomes. None of, or only a small number of, our R&D programs may actually result in the commercialization of a product. We will not be able to commercialize our products if preclinical studies do not produce successful results or if clinical trials do not demonstrate safety and efficacy in humans.
Success in preclinical studies or early-stage clinical trials does not ensure that later stage clinical trials will be successful, nor does it ensure that regulatory clearance or approval for the product will be obtained. In addition, the process for the completion of preclinical and clinical trials is lengthy and may be subject to a number of delays for various reasons, which would delay the commercialization of any successful product. If our development projects are not successful or are significantly delayed, we may not recover our substantial investments in the product and our failure to bring these products to market on a timely basis, or at all, could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our Common Stock to decline.
Obtaining regulatory clearances and approvals for new products and manufacturing processes can take a number of years and involve the expenditure of substantial resources. Despite the substantial time and expense invested, regulatory clearance or approval is never guaranteed. The number, size and design of clinical trials that will be required will vary depending on the product or condition for which the product is intended to be used and the regulations and guidance documents applicable to any particular product. Additionally, during the review process and prior to approval, the FDA or other regulatory bodies could require additional data, which could delay approval. The FDA or other regulators can delay, limit or deny clearance or approval of a product for many reasons, including governmental resources availability and allocation, or adopt new policies or regulations requiring new or different evidence of safety and efficacy for the intended use of a product. Further, given changes to the U.S. government’s policies and priorities since the current presidential administration entered office in January 2025, there is substantial uncertainty as to how, if at all, the administration will seek to modify or revise the requirements and policies of the FDA and other regulatory agencies. There is also uncertainty as to how other measures being implemented by the current administration across the government will impact our activities and those of the FDA and its operations. Over the last several years, the U.S. government has experienced prolonged shutdowns and certain regulatory agencies, such as the FDA, have had to furlough employees and halt critical activities. If a government shutdown were to occur, it could significantly impact the ability of agencies to timely review and process our regulatory submissions, which would have a material adverse effect on our business.
In addition, even if such clearance or approval is secured, the approved labeling may have significant labeling limitations, including limitations on the indications for which we can market a product, or require onerous risk management programs. Furthermore, from time to time, changes to the applicable legislation, regulations or policies may be introduced that change these review and approval processes for our products, which may make it more difficult and costly to obtain or maintain regulatory clearances and approvals.
Successful results in clinical trials and in the subsequent application for marketing approval are not guaranteed. If we are unable to obtain regulatory clearances and approvals, we will not be able to commercialize and generate revenue from our products. Even if we receive regulatory clearance or approval for any of our products, our profitability will depend on our ability to commercialize and generate revenues from their sale or the licensing of our technology. The failure to commercialize our products could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our Common Stock to decline.
Even if a product receives regulatory clearance or approval, it may still face development and regulatory difficulties that could delay or impair future sales of products.
Following initial regulatory clearance or approval of any products, we will be subject to continuing regulatory review by various government authorities in those countries where our products are marketed or intended to be marketed, including the review of potential malfunctions and adverse events that are reported after products become commercially available. In addition, we will be subject to ongoing audits and inspections of our facilities and products by the FDA, as well as other regulatory agencies in and outside the United States. Previously unknown problems with the product could result in restrictions on the marketing of the product, including withdrawal of the product from the market.
In addition, if we were to receive regulatory clearance or approval to sell our DurAVR® THV System or another product, the relevant regulatory authorities could, nevertheless, impose significant restrictions on the indicated uses, manufacturing, labelling, packaging, adverse event reporting, storage, advertising, promotion and record keeping or impose ongoing requirements for post-approval studies.
If we fail to comply with the regulatory requirements in those countries where our products are sold, we could lose our marketing clearances or approvals or be subject to fines or other sanctions. Also, as a condition to granting marketing clearance or approval of a product, the applicable regulatory agencies may require a company to conduct additional clinical trials or remediate cGMP issues, the results of which could result in the subsequent loss of marketing approval, changes in product labeling or new or increased concerns about side effects or efficacy of a product.
In addition, incidents of medical device related adverse events or unintended side effects or misuse relating to our products could result in additional regulatory controls or restrictions or lead to a recall or withdrawal of the product from the market. A recall or market withdrawal, whether voluntary or required by a regulatory authority, may involve significant costs to us, potential disruptions in the supply of our products to our customers and reputational harm to our products and business, all of which could harm our ability to market our products and could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our Common Stock to decline.
Even with regulatory clearance or approval to bring a product to market, our profitability may be impacted by ongoing coverage and reimbursement determinations by government health care programs and other third-party payors for our products, or procedures and services that rely on our products.
Our products and technologies may be paid for by the CMS and other government or third-party payors or will be used by hospitals and health care providers who are reimbursed for procedures and services involving our products. Such payment determinations are subject to pre-approval qualifications and satisfaction of appropriate criteria. CMS, or other third-party payors, may seek to lower costs or limit use of our products as a means to achieve lower health care costs. The sale and demand for our products may be adversely impacted by such coverage and reimbursement determinations.
Participation in government health care programs and contracts with third-party payors will require ongoing compliance with federal and state health care laws and agreement terms.
We will be subject to ongoing monitoring for compliance with federal and state laws, as well as contractual terms, if we receive third-party payor reimbursement for our products, or are engaged with entities that receive reimbursement for procedures and services involving our products. Violation of such laws or contractual terms may result in significant fines and fees, withholding of payment, or removal from the third-party payor programs, which would impact our profitability.
Our products that are in development may not achieve market acceptance, if approved, which could limit our growth and adversely affect our business, financial condition and results of operations.
Even if the FDA or any comparable foreign regulatory authority clears or approves the marketing of any product that we develop, physicians, healthcare providers, patients or the medical community may not accept or use them. DurAVR® THV System and other products are still in the development stage and are based on our proprietary technologies. We do not have proven marketing or sales strategies for such new products, nor do we know if customers will accept our products, if approved, and therefore we do not know how the introduction of any approved products will affect our business. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenues or any profits from operations. Our product portfolio continues to expand, and we are investing significant resources to enter into, and in some cases create new markets for, our products. We are continuing to invest resources to achieve clearance and approval and market acceptance of our products but are unable to guarantee that we will succeed.
The degree of market acceptance of our products, if approved, will depend on a number of factors, including:
•
the timing of market introduction of our products, as well as competitive products;
•
the clinical indications for which a product is approved;
•
perceived benefits from our products;
•
perceived cost effectiveness of our products;
•
perceived safety and effectiveness of our products;
•
the effectiveness of sales and marketing efforts;
•
the terms of any clearances or approvals and the countries in which clearances and approvals are obtained;
•
our ability to provide acceptable evidence of safety and efficacy;
•
marketing, manufacturing and supply support;
•
potential product liability claims;
•
the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors and government authorities;
•
in certain instances, reimbursement available through government and private healthcare programs for using our products; and
•
introduction and acceptance of competing products or technologies.
If our products do not gain market acceptance or if our customers prefer our competitors’ products, our potential revenue growth would be limited, which would adversely affect our business, financial condition and results of operations. Even if some of our products achieve market acceptance, the market may not prove to be large enough to allow us to generate significant revenues.
Failure to successfully innovate and develop new and differentiated products in a timely manner and effectively market these products could have a material effect on our prospects.
Our continued growth and success depend on our ability to innovate and develop new and differentiated products in a timely manner and effectively market these products. Without the timely innovation and development of products, our products could be rendered obsolete or less competitive because of the introduction of a competitor’s newer technologies. Innovating products requires the devotion of significant financial and other resources to R&D activities; however, there is no certainty that the products we are currently developing will complete the development process, or that we will obtain the regulatory or other clearances or approvals required to market such products in a timely manner or at all. Even if we timely innovate and develop products, our ability to successfully market them could be constrained by a number of different factors, including competitive products and pricing, barriers in patients’ treatment pathway, the need for regulatory clearance or approval, restrictions imposed on cleared or approved indications, and uncertainty over third-party reimbursement. Failure in any of these areas could have a material effect on our prospects.
We may find it difficult to enroll patients in our clinical trials, and patients could discontinue their participation in clinical trials, which could delay or prevent clinical trials and make those trials more expensive to undertake.
Identifying and qualifying patients to participate in current and future clinical trials of our products is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our products. Patients could be unavailable for various reasons, including competitive clinical trials for similar patient populations, eligibility criteria for the clinical trial, and the proximity of patients to clinical sites. In addition, the process of identifying, confirming eligibility and enrollment of patients may prove costly, and there is a risk that patients enrolled in clinical trials will drop out of the trials before the administration of our products or trial completion. As such, the timeline for recruiting patients, conducting trials and obtaining regulatory clearance or approval of products may be delayed. If we have difficulty enrolling a sufficient number of patients to conduct any clinical trials as planned or maintaining such enrollment, we may need to delay, limit or discontinue those clinical trials. Clinical trial delays could result in increased costs, slower product development, setbacks in testing the safety and effectiveness of our technology or discontinuation of the clinical trials altogether. In addition, some of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the delay or denial of regulatory clearance or approval of our products.
Interim, top-line and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or top-line data from our preclinical studies and clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the top-line or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, top-line data should be viewed with caution until the final data are available.
From time to time, we may also disclose interim data from our preclinical studies and clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available or as patients from our clinical trials continue other treatments for their disease. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
Others, including regulatory authorities, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically a significant volume of data and other information, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure. If the interim, top-line, or preliminary data that we report differ from final results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain clearance or approval for, and commercialize, our products may be harmed, which could harm our business, operating results, prospects or financial condition.
We operate in a highly competitive and rapidly changing industry, and if we do not compete effectively, our business will be harmed.
The medical technology industry is highly competitive and subject to significant and rapid technological change. Our success is highly dependent on our ability to discover, develop and obtain regulatory clearance or approval for new and innovative products on a cost-effective basis and to market them successfully. In doing so, we face and will continue to face intense competition from a variety of businesses, including large healthcare companies, academic institutions, government agencies and other public and private research organizations. These organizations may have significantly greater resources than we do and conduct similar research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and marketing of products that compete with our products. Mergers and acquisitions in the medical technology industry may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries.
We expect to face increasingly intense competition as new technologies become available. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Any products that we successfully develop and commercialize will compete with existing products and new products that may become available in the future. The highly competitive nature of, and rapid technological changes in the medical technology industry could render our products or our technology obsolete, less competitive or uneconomical. Our competitors may, among other things:
•
have significantly greater financial, manufacturing, marketing, development, technical and human resources than we do;
•
develop and commercialize products that are safer, more effective, less expensive, easier to implement or have fewer or less severe side effects;
•
obtain quicker regulatory clearance or approval;
•
establish superior proprietary positions covering our products and technologies;
•
implement more effective approaches to sales and marketing; or
•
form more advantageous strategic alliances.
Should any of these factors occur, our business, financial condition and results of operations could be materially adversely affected. Competing products could present superior alternatives, including by being more effective, safer, less expensive or marketed and sold more effectively than any products we may develop. Competitive product approaches may make any products we develop obsolete or non-competitive before we recover the expense of developing and commercializing our products.
Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
The success of many of our products may depend upon the knowledge and experience of certain key physicians and heart valve centers.
We work with leading global physicians who form our Global Medical Advisory Board, which provides guidance to us on building clinical validation of the DurAVR® THV System. These physicians provide considerable knowledge and experience. These physicians may assist us as researchers, marketing consultants, product trainers and consultants and as public speakers. If new laws or other developments limit our ability to appropriately engage these professionals or with the heart valve centers of which they are a part or to continue to receive their advice and input or we are otherwise unsuccessful in maintaining strong working relationships with these physicians or their heart valve centers, then the development, marketing and use of our products could suffer, which could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Operations
Our operating results could be adversely affected if we are unable to accurately forecast demand for any of our products that receive marketing clearance or approval and if we are unable to adequately manage our inventory.
If one or more of our products receives marketing clearance or approval, and we commercialize the product, to ensure adequate inventory supply, we will be required to forecast inventory needs and expenses and place orders sufficiently in advance with our suppliers and contract manufacturers, based on our estimates of future demand for our products. Failure to accurately forecast our needs could result in manufacturing delays or increased costs. Due to the lead times necessary to obtain and install new equipment and ramp up production of product lines, if we fail to adequately forecast the need for additional manufacturing capacity, we may be unable to scale production in a timely manner to meet demand for our products. In addition, the technically complex manufacturing processes required to manufacture our products increase the risk of production failures and can increase the cost of producing our products. As a result, because the production process for our products is complex and sensitive, the cost of production and the chance of production failures and lengthy supply interruptions is increased, which can have a substantial impact on our inventory levels.
Our ability to accurately forecast demand could be affected by many factors, including changes in demand for our products, changes in demand for the products of our competitors and the weakening of economic conditions or confidence in future economic conditions. This risk could be exacerbated by the fact that we may not carry a significant amount of inventory and may not be able to satisfy short-term demand increases, or at times will have an excess in inventory that we are unable to effectively utilize. If we fail to accurately forecast demand, we could experience excess inventory levels or a shortage of products available for sale and any such shortage could have a material impact on our business operations.
The expansion of our manufacturing capabilities may be unsuccessful.
We have been manufacturing the ADAPT® tissue for many years. However, to continue the development of our current and future products, we will need to expand our manufacturing capabilities, including potentially outsourcing specific manufacturing processes. Problems with expansion of our manufacturing capabilities, including issues with third-party manufacturers, could delay clinical trials and the commercialization of our products, if approved.
We rely on third parties to conduct our clinical trials and preclinical studies. If these third parties do not successfully carry out their contractual duties, comply with applicable regulatory requirements or meet expected deadlines, our development programs and our ability to seek or obtain regulatory clearance and approval for or commercialize our products may be delayed.
We are dependent on third parties to conduct our clinical trials and preclinical studies for our DurAVR® THV System. Specifically, we rely on, and will continue to rely on, medical institutions, clinical investigators, lab service providers, and consultants to conduct clinical trials and preclinical studies, in each case in accordance with trial protocols and regulatory requirements. These third parties play a significant role in the conduct, monitoring, project and site management, data management, safety and lab services of our trials studies, including subsequent analysis of data. Though we expect to carefully manage our relationships with such third parties, there can be no assurance that we will not encounter challenges or delays in the future, or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects. Furthermore, while we have and will have agreements governing the activities of our third-party contractors, we have limited influence over their actual performance. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards and requirements, and our reliance on third parties does not relieve us of our regulatory responsibilities.
While the third parties upon which we rely change from time to time and for each study, these partners include:
•
Bright Research, which is a clinical research organization that provides us with clinical data monitoring, project and site management, data management, and safety reporting services;
•
CRF, which provides us with core lab services and an independent clinical events committee; and
•
QMED, which provides clinical trial support for the EU, including the provision of life science services in the areas of regulatory affairs, training, quality assurance and control, clinical trial consultancy and submission support to the EU authorities.
In addition, we and the third parties we work with are required to comply with Good Laboratory Practice (“GLP”) and Good Clinical Practice (“GCP”) requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities. Furthermore, our clinical trials must be conducted with materials manufactured in accordance with cGMP regulations. Regulatory authorities enforce GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of the third parties we work with or our trial sites fail to comply with applicable GLP, GCP or other requirements, the data generated in our preclinical studies or clinical trials may be deemed unreliable, and the FDA or comparable foreign regulatory authorities may require us to perform additional studies or trials before approving our marketing applications, if ever. Furthermore, our clinical trials must be conducted with materials manufactured in accordance with cGMP regulations. Failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and our goal of receiving premarket approval for a product.
There is no guarantee that any of the third parties with whom we work will devote adequate time and resources to such trials or studies or perform as contractually required. If any of these third parties fails to meet expected deadlines, adhere to our clinical protocols or meet regulatory requirements or otherwise perform in a substandard manner, our clinical trials may be extended, delayed or terminated. In addition, the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other activities that could harm our competitive position.
In addition, the third parties with whom we work have the right to terminate their agreements with us in the event of an uncured material breach and under other specified circumstances. If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties on commercially reasonable terms, in a timely manner or at all. Switching or adding additional third parties involves additional cost and requires our management’s time and focus. In addition, there is a natural transition period when a new third-party service provider commences work. As a result, delays can occur, which can materially impact our ability to meet our desired clinical development timelines. Though we work to carefully manage our relationships with the third parties with whom we work, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
We rely on third parties for the supply of materials and for the design and manufacture of our products. Any failure by or loss of a vendor could result in delays and increased costs, which may adversely affect our business.
We currently rely on a limited number of suppliers, including several single-source suppliers, to supply raw materials and other components and on contract manufacturers to design and manufacture certain products. The facilities used by our contract manufacturers must be approved for the manufacture of our products by the FDA, or any comparable foreign regulatory authority, pursuant to inspections that may be conducted by or for regulatory authorities. We do not control the manufacturing process of, and are completely dependent on, contract manufacturers for compliance with cGMP requirements for manufacture of those products. If these contract manufacturers cannot successfully manufacture such products in a manner that conforms to our specifications and the strict regulatory requirements of the FDA or any comparable foreign regulatory authority, they will not be able to secure and/or maintain regulatory approval for the use of their manufacturing facilities.
We also purchase certain supplies and services from single sources for reasons of quality assurance, cost-effectiveness, availability, constraints resulting from regulatory requirements and other reasons. We experience from time to time, and may continue to experience, supply interruptions due to a variety of factors, including:
•
general economic conditions that could adversely affect the financial viability of our vendors;
•
vendors’ election to no longer service or supply medical technology companies, including due to the burdens of applicable quality requirements and regulations or for no reason at all;
•
the limitation or ban of certain chemicals or other materials used in the manufacture of our products; and
•
delays or shortages due to trade or regulatory embargoes.
Additionally, any significant increases in the cost of raw materials, whether due to inflationary pressure, supply constraints or regulatory changes could adversely impact our operating results. A change or addition to our vendors could require significant effort due to the rigorous regulations and requirements of the FDA and other regulatory authorities. It could be difficult to establish additional or replacement sources on a timely basis or at all, which could have a material adverse effect on our business. See the section entitled “Business – Single Source Suppliers” for a description of the agreements we are party to with our single-source suppliers.
We have limited control over our suppliers, contract manufacturers, and logistic partners, and such limited control could subject us to significant risks, including the potential inability to produce or obtain quality products and services on a timely basis or in sufficient quantity.
We currently rely on a limited number of suppliers to supply raw materials and other components for certain of our products, contract manufacturers to manufacture certain of our products, and logistics partners to transport certain of our products. We have limited control over our suppliers, contract manufacturers and logistics partners. Such limited control could subject us to the following risks:
•
inability to satisfy demand for our current and future products and services;
•
reduced control over delivery timing and related customer experience and product reliability;
•
reduced ability to monitor the manufacturing process and components used in our products;
•
limited ability to develop comprehensive manufacturing specifications that take into account any materials shortages or substitutions;
•
variance in the manufacturing capability of our third-party manufacturers;
•
price increases;
•
failure of a significant supplier or manufacturer partner to perform its obligations to us for technical, market or other reasons;
•
variance in the quality of services provided by our third-party partners;
•
inability of suppliers to comply with applicable provisions of the FDA’s Device cGMP or other applicable laws enforced by the FDA, state regulatory authorities or non-United States regulatory authorities;
•
inability to ensure the quality of products and components manufactured by third parties;
•
production delays related to the evaluation and testing of products and components from alternative suppliers and corresponding regulatory qualifications;
•
difficulties in establishing additional supplier or manufacturer partner relationships if we experience difficulties with our existing suppliers, manufacturers or logistics partners;
•
shortages of materials or components;
•
production shortages resulting from any events affecting raw material supply;
•
misappropriation of our intellectual property;
•
exposure to natural catastrophes, epidemics such as a pandemic, political unrest, terrorism, labor disputes and economic instability resulting in the disruption of trade from foreign countries in which our products or the components are sourced;
•
changes in local economic conditions in the jurisdictions where our suppliers, manufacturers, and logistics partners are located;
•
the imposition of new laws, including those relating to labor conditions, quality and safety standards, imports, duties, tariffs, taxes and trade restrictions; and
•
insufficient warranties and indemnities on components supplied to our manufacturers or performance by our partners.
If our suppliers became unable to provide components in the volumes needed or at an acceptable price, we would have to identify and qualify acceptable replacements from alternative sources of supply. The process of qualifying suppliers is lengthy. Delays or interruptions in the supply of our requirements could limit or stop our ability to provide sufficient quantities of devices on a timely basis or meet demand for our devices, which could have a material adverse effect on our business, financial condition and results of operations.
Furthermore, our failure or the failure of our manufacturing partners and suppliers to maintain compliance with the applicable regulatory requirements could result in the shutdown of our manufacturing operations or the recall of our products, which would harm our business. In the event that one of our manufacturing partners or suppliers fails to maintain compliance with our or governmental quality requirements, we may have to qualify a new manufacturing partner or supplier, and we could experience manufacturing delays as a result.
The occurrence of any of these risks could cause us to experience a significant disruption in our ability to produce and deliver our products to our customers and could harm our brand and reputation.
Health and safety hazards may adversely affect our business operations.
We have been engaged in manufacturing and R&D activities for a number of years. Our manufacturing and R&D activities are conducted within our premises in Australia and the United States. In light of our business, there are health and safety risks that our employees and contractors could be exposed to. Such health and safety risks include all hazards and risks related to work activities, including both physical and mental health risks. There is a heightened level of risk in a manufacturing environment but health and safety risks also arise in R&D facilities as well as office environments. They may arise due to insufficiently trained or qualified personnel, equipment failure, staff fatigue, unsafe work environments and/or deficient health and safety management systems.
Health and safety incidents in the workplace could directly impact staff, including injury or fatality, mental health and operational performance. It could also result in an increase in litigation and insurance claims, reputational impacts and regulatory intervention. Thus, any health and safety incident occurring to our employees and contractors could materially affect our business operations.
Our R&D efforts will be jeopardized if we are unable to retain key personnel and cultivate key academic and scientific collaborations.
Changes in our senior management can be disruptive to our business and may adversely affect our operations. For example, when we have changes in senior management positions, we may elect to adopt different business strategies or plans. Any new strategies or plans, if adopted, may not be successful and if any new strategies or plans do not produce the desired results, our business may suffer.
Moreover, competition for qualified employees is intense and as such we may not be able to attract and retain personnel critical to our success. Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel, manufacturing personnel, sales and marketing personnel and on our ability to develop and maintain important relationships with clinicians, scientists and leading academic and health institutions. Given the specialized nature of our products, there is an inherent scarcity of experienced personnel in these fields. As we continue developing products in our pipeline, we will require personnel with medical, scientific or technical qualifications specific to each program. The loss of key personnel, in particular our senior leadership team, could delay our R&D activities. Despite our efforts to retain valuable employees, members of our team may terminate their employment with us on short notice. The competition for qualified personnel in the medical technology industry is intense, and our future success depends upon our ability to attract, retain and motivate highly skilled scientific, technical and managerial employees. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our product development and commercialization activities.
We may in the future seek to identify and acquire certain assets, products and businesses, and there can be no guarantee that we will be able to successfully consummate such transactions.
We may in the future seek to identify and acquire complementary businesses, products, technologies or other assets to augment our pipeline. Such transactions may be complex, time consuming and expensive. There can be no guarantee that we will be able to successfully consummate acquisitions or other arrangements, which could result in significant diversion of management and other employee time, as well as substantial out-of-pocket costs. If such transactions are not completed for any reason, we may incur significant costs and the market price of our Common Stock may decline.
In addition, even if an acquisition is consummated, the integration of the acquired business, product or other assets into our company may be complex and time-consuming, and we may not achieve the anticipated benefits, cost-savings or growth opportunities we expect. Potential difficulties that may be encountered in the integration process include the following: integrating personnel, operations and systems, while maintaining focus on selling and promoting existing and newly-acquired products; coordinating geographically dispersed organizations; preventing the distraction of management and employees from operations; retaining existing customers and attracting new customers; maintaining the business relationships the acquired company has established, including with health care providers; and managing inefficiencies associated with integrating the operations of our company and the acquired business, product or other assets.
To the extent we are able to enter into collaborative arrangements or strategic alliances, we will be exposed to risks related to those collaborations and alliances.
The rapid pace of technological development in the medical technology industry and the specialized expertise required in different areas of medicine make it difficult for one company alone to develop a broad portfolio of technological solutions. In addition to internally generated growth through our R&D efforts. We expect to make future investments where we believe that we can stimulate the development or acquisition of new technologies and products to further our strategic objectives and strengthen our existing businesses. Collaborative arrangements and strategic alliances in and with medical technology companies are inherently risky, and we cannot guarantee that any of our previous or future investments or investment collaborations will be successful or will not materially adversely affect our business, results of operations, financial condition and cash flows.
Any collaboration arrangement or alliance we have or may have in the future could be terminated for reasons beyond our control or we may not be able to negotiate future alliances on acceptable terms, if at all. These arrangements and alliances could result in us receiving less revenue than if we sold our products directly, place the development, sales and marketing of our products outside of our control, require us to relinquish important rights or otherwise be on unfavorable terms.
Collaborative arrangements or strategic alliances will also subject us to a number of risks, including the risk that:
•
we may not be able to control the amount and timing of resources that our strategic partners/collaborators may devote to the products;
•
strategic partners/collaborators may experience financial difficulties;
•
the failure to successfully collaborate with third parties may delay, prevent or otherwise impair the development or commercialization of our products or revenue expectations;
•
business combinations or significant changes in a collaborator’s business strategy may adversely affect a collaborator’s willingness or ability to complete their obligations under any arrangement;
•
a collaborator could independently move forward with a competing product developed either independently or in collaboration with others, including our competitors; and
•
collaborative arrangements are often terminated or allowed to expire, which would delay the development of, and may increase the cost of developing, products.
We are subject to various risks relating to international activities that could affect our overall profitability.
Our international operations subject us to a number of risks, which may vary significantly from the risks we face in our United States operations, including:
•
fluctuations in currency exchange rates;
•
domestic and global economic conditions such as inflation or recession;
•
healthcare legislation and other regulations;
•
differing standards and privacy requirements for the conduct of clinical trials;
•
differing procedures and standards for regulatory approval and commercialization;
•
tariffs and other trade barriers;
•
compliance with foreign medical device manufacturing regulations;
•
challenges with obtaining required supplies of components for our devices;
•
difficulty in enforcing agreements and collecting receivables through foreign legal systems;
•
reduction in third-party payor reimbursement for our products;
•
inability to obtain import licenses;
•
the impact from health epidemics/pandemics on the global economy;
•
the impact of geopolitical tensions and/or conflicts, including the war in Ukraine;
•
changes in trade policies and in United States and foreign tax policies;
•
possible changes in export or import restrictions;
•
differing labor regulations and difficulty in staffing and managing foreign operations;
•
the modification or introduction of other governmental policies with potentially adverse effects; and
•
limitations on our ability under local laws to protect our intellectual property.
We are subject to risks associated with currency fluctuations and changes in foreign currency exchange rates could impact our results of operations.
If the Australian dollar weakens against the United States dollar, then, if we decide to convert our Australian dollars into United States dollars for any business purpose, appreciation of the United States dollar against the Australian dollar would have a negative effect on the United States dollar amount available to us. To the extent that we need to convert United States dollars we receive into Australian dollars for our operations, appreciation of the Australian dollar against the United States dollar would have a negative effect on the Australian dollar amount we would receive from the conversion. Consequently, appreciation or depreciation in the value of the Australian dollar relative to the United States dollar would affect our financial results. As a result of such foreign currency fluctuations, it could be more difficult to detect underlying trends in our business and results of operations.
Any failure to protect our information technology infrastructure and our products against cyber-based attacks, network security breaches, service interruptions, artificial intelligence or data corruption could materially disrupt our operations and adversely affect our business and operating results.
The operation of our business depends on our information technology systems. We rely on our information technology systems to effectively manage sales and marketing data, accounting and financial functions, inventory management, product development tasks, clinical data, customer service and technical support functions. Our information technology systems are vulnerable to damage or interruption from earthquakes, fires, floods and other natural disasters, terrorist attacks, power losses, computer system or data network failures, security breaches and data corruption.
In addition, our information technology infrastructure and products are vulnerable to cyber-based attacks. Cyber-based attacks can include computer viruses, denial-of-service attacks, phishing attacks, ransomware attacks and other introduction of malware to computers and networks; unauthorized access through the use of compromised credentials; exploitation of design flaws, bugs or security vulnerabilities; intentional or unintentional acts by employees or other insiders with access privileges; and intentional acts of vandalism by third parties and sabotage. Further, cybersecurity threats and the techniques used in cyber-based attacks change, develop, and evolve rapidly, including from emerging technologies, such as advanced forms of artificial intelligence (“AI”) and quantum computing. In addition, laws of applicable jurisdictions can expose us to investigations and enforcement actions by regulatory authorities and claims from individuals potentially resulting in penalties and significant legal liability if our information technology security efforts are inadequate.
Significant disruption in either our or our service providers’ or suppliers’ information technology could impede our operations or result in decreased sales, result in liability claims or regulatory penalties, or lead to increased overhead costs, product shortages, loss or misuse of proprietary or confidential information, intellectual property, or sensitive or personal information, all of which could have a material adverse effect on our reputation, business, financial condition and operating results.
Artificial intelligence technologies could present business, compliance and reputational risks.
Recent technological advances in AI and machine-learning technology both present opportunities and pose risks to us. If we fail to keep pace with rapidly evolving technological developments in AI, our competitive position and business results may suffer. We face risk of competitive disadvantage if our competitors more effectively use AI to better serve customers, drive internal efficiencies, and/or create new or enhanced products or services. We have begun to incorporate AI capabilities into our operations and the introduction of these technologies, particularly generative AI, into internal processes, customer engagements, and/or new and existing product offerings may result in new or expanded risks and liabilities, including due to enhanced governmental or regulatory scrutiny, litigation, compliance issues, ethical concerns, confidentiality or security risks, as well as other factors that could adversely affect our business, reputation, and financial results. In addition, our personnel could, unbeknownst to us, improperly utilize AI and machine learning-technology while carrying out their responsibilities. The use of AI in the development of our products and services could also cause loss of intellectual property, as well as subject us to risks related to intellectual property infringement or misappropriation, data privacy and cybersecurity. The use of AI can lead to unintended consequences, including generating content that appears correct but is factually inaccurate, misleading or otherwise flawed, or that results in unintended biases and discriminatory outcomes, which could harm our reputation and business and expose us to risks related to inaccuracies or errors in the output of such technologies. Finally, multiple jurisdictions have either already put in place laws and regulations governing the use of AI, or are considering such laws and regulations. Compliance with these laws, regulations, and industry frameworks may limit our ability to leverage AI or require us to substantially revise our approach to its use.
Our business, data, services and products are or may become subject to United States federal and state and international data privacy laws and regulations and any failure to comply with these laws and regulations could harm our reputation, expose us to damages and otherwise adversely affect our business.
As a global company, we are or may become subject to laws and regulations in the United States and other countries concerning the handling of personal data, including but not limited to those related to the collection, storage, handling, use, disclosure, transfer, and security of personal data. These laws and regulations include, for example, the European Union’s General Data Protection Regulation and the California Consumer Privacy Act, and other similar United States state privacy laws. These laws and regulations are continuously evolving and developing, creating significant uncertainty as privacy and data protection laws may be interpreted and applied differently from country to country and may create inconsistent or conflicting requirements. Our compliance with privacy and data protection laws may result in significant costs and challenges that are likely to increase over time. Any failure, or perceived failure, by us or third-party service providers to comply with our privacy or security policies or privacy-related legal obligations may result in governmental enforcement actions, litigation, or negative publicity, and could have an adverse effect on our operating results and financial condition
Increased emphasis on environmental, social, and governance (“ESG”) matters may have an adverse effect on our business, financial condition, results of operations and reputation.
Investors, regulators, legislators, customers, consumers, employees, and other key stakeholders are increasingly focusing on areas of corporate responsibility, and particularly matters related to ESG factors. Such matters could include, among other things, environmental stewardship, diversity, equity, and inclusion initiatives, supply chain practices, good corporate governance, workplace conduct, and support for local communities. Institutional investors have expressed expectations with respect to ESG matters that they use to guide their investment strategies and may, in some cases, choose not to invest in us if they believe our ESG policies are lagging or inadequate. Other stakeholders also have expectations regarding ESG factors, such as employees or potential employees who desire to work for a company that reflects their personal values. These areas of focus are continuing to evolve, as are the criteria on which investors assess companies’ performance in these areas. Certain investors are increasingly looking to companies that demonstrate strong ESG and sustainability practices as an indicator of long-term resilience. Keeping up with and meeting these expectations may disrupt our business and divert the attention of our management, and we may be unable to make the investments in ESG programs that our competitors with greater financial resources are able to make. Failure to meet the expectations of investors and other stakeholders in these areas may damage our reputation, impact employee retention, impact the willingness of our customers to do business with us, or otherwise impact our financial results and stock price.
Risks Related to Legal and Regulatory Matters
We could become exposed to product liability claims that could harm our business and we may be unable to obtain insurance coverage at acceptable costs and adequate levels.
The clinical trials and sales of medical products entails an inherent risk of product liability. We rely on a number of third-party researchers and contractors to produce, collect, and analyze data regarding the safety and efficacy of our products. We also have quality control and quality assurance in place to mitigate these risks and have historically obtained professional liability and clinical trial insurance on our clinical trials to cover financial damages in the event that human testing is done incorrectly, or the data is analyzed incorrectly.
Notwithstanding our control procedures, we could face product liability exposure related to the testing of our products in clinical trials. If any of our products are approved for sale, we could face exposure to claims by an even greater number of persons than were involved in the clinical trials once marketing, distribution and sales of our products begin.
Regardless of merit or eventual outcome, liability claims could result in:
•
decreased demand for our products;
•
injury to our reputation;
•
withdrawal of clinical trial participants;
•
costs of related litigation;
•
substantial monetary awards to patients and others;
•
loss of revenues; and
•
the inability to commercialize products.
If a claim is made against us in conjunction with these research testing activities, the market price of our Common Stock could be negatively affected.
Use of our products in unapproved circumstances could expose us to liabilities.
The marketing approvals, if any, from the FDA and other regulators of certain of our products are expected to be limited to specific indications. Such approvals would prohibit us from marketing or promoting any unapproved use of our products. Physicians, however, can use these products in ways or circumstances other than those strictly within the scope of the regulatory approval. Although we intend that the product training we will provide to physicians and other healthcare professionals will be conducted in compliance with applicable laws, and therefore, will be mainly limited to approved uses or for clinical trials, no assurance can be given that claims might not be asserted against us if our products are used in ways or for procedures that are not approved.
Disputes could substantially disrupt our business operations.
Even if resolved in our favor, litigation or other legal proceedings commenced against us by stockholders, regulatory authorities, employees, competitors or other third parties could cause us to incur significant expenses and could distract our personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and, if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our Common Stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to develop our products, continue our internal research programs or enter into strategic collaborations that could help us bring our products to market. As a result, uncertainties resulting from the initiation and continuation of litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
Our products and operations are subject to extensive government regulation, including environmental, health and safety regulations, which could result in substantial costs and any failure to comply with applicable requirements could harm our business.
Our medical devices are subject to rigorous regulation and scrutiny by the FDA and other governmental authorities. Government regulation applies to nearly all aspects of our products’ lifecycles, including testing, clinical study, manufacturing, transporting, sourcing, safety, labeling, storing, packaging, recordkeeping, reporting, advertising, promoting, distributing, marketing, and importing or exporting of medical devices and products. In general, unless an exemption applies, a medical device or product must receive regulatory clearance or approval or clearance before it can be marketed or sold. Modifications to existing products or the marketing of new uses for existing products also may require regulatory clearance or approvals, or supplemental approvals. If we are unable to obtain these required marketing authorizations, our ability to commercialize new products will be delayed or adversely impacted.
Regulatory agencies may refuse to grant approval or clearance or disagree with our interpretation of the data, or disagree with our interpretation of the regulatory requirements, such as products that are subject to enforcement discretion or consumer products that do not meet the definition of an FDA-regulated medical device. Furthermore, the FDA and other regulatory agencies could change their policies, adopt additional regulations, or revise existing regulations, or change regulatory and policy priorities, each of which could impact how our products are regulated, prevent or delay approval or clearance of devices, or impact our ability to market a previously cleared, approved, or unregulated device. Our failure to comply with regulatory requirements of the FDA or other applicable regulatory requirements in the United States or elsewhere could subject us to administratively or judicially imposed sanctions. These sanctions could include warning letters, fines, civil penalties, criminal penalties, injunctions, debarment, product seizure or detention, product recalls and total or partial suspension of production, sale and/or promotion. Any of the foregoing actions could have a material adverse effect on our financial condition and results of operations. In addition to any such sanctions for noncompliance described above, commencement of an enforcement proceeding, inspection or investigation could divert substantial management attention from the operation of our business and, as a result, have an adverse effect on our business.
Our operations are subject to environmental, health, and safety regulations that could result in substantial costs.
Our operations are subject to environmental, health, and safety laws, and regulations concerning, among other things, the generation, handling, transportation, and disposal of hazardous substances or wastes, the cleanup of hazardous substance releases, and emissions or discharges into the air or water. We may incur in the future expenditures in connection with environmental, health and safety laws, and regulations. New laws and regulations, violations of these laws or regulations, stricter enforcement of existing requirements, or the discovery of previously unknown contamination could require us to incur costs or could become the basis for new or increased liabilities that could be material.
Healthcare policy changes may have a material adverse effect on us.
There have been and continue to be actions and proposals by several governments, regulators and third-party payers globally, including the United States federal and state governments, to control healthcare costs and, more generally, to reform healthcare systems. The healthcare industry in the United States is subject to fundamental changes due to ongoing federal and state healthcare reform efforts and related political, economic, and regulatory influences, including those from the recent change in presidential administration. Certain of these actions and proposals, among other things, limit the prices we are able to charge for our products or the amounts of reimbursement available for our products, increase the importance of our ability to compete on cost, and could limit the acceptance and availability of our products. These actions and proposals could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We could be exposed to significant liability claims if we are unable to obtain insurance at acceptable costs and adequate levels or otherwise protect ourselves against potential product liability claims.
The design, manufacture and marketing of medical device products involve certain inherent risks. Manufacturing or design defects, unanticipated use of our products, or inadequate disclosure of risks relating to the use of our products could lead to negative publicity, government investigation, litigation or other adverse events. These events could lead to recalls or safety alerts relating to our products (either voluntary or required by the FDA, or similar governmental authorities in other countries) and could result, in certain cases, in the removal of a product from the market. A recall could result in significant costs, as well as negative publicity and damage to our reputation that could reduce demand for our products. In some circumstances, such adverse events could also cause delays in new product clearance and commercialization plans.
The testing, manufacture, marketing and sale of medical devices entail the inherent risk of liability claims or product recalls. Product liability insurance is expensive and, if available, may not be available on acceptable terms at all periods of time. A successful product liability claim or product recall could inhibit or prevent the successful commercialization of our products, cause a significant financial burden on us, or both, which in either case could have a material adverse effect on our business and financial condition.
We are subject to various United States and international bribery, anti-kickback, false claims, privacy, transparency, and similar laws, any breach of which could cause a material, adverse effect on our business, financial condition, and profitability.
Our relationships with physicians, hospitals, and other healthcare providers are subject to scrutiny under various United States and international bribery, anti-kickback, false claims, privacy, transparency, and similar laws, often referred to collectively as “healthcare compliance laws.” Healthcare compliance laws are broad, sometimes ambiguous, complex, and subject to change and changing interpretations. Possible sanctions for violation of these healthcare compliance laws include fines, civil and criminal penalties, exclusion from government healthcare programs, and despite our compliance efforts, we face the risk of an enforcement activity or a finding of a violation of these laws. While our relationships with healthcare professionals and organizations are structured to comply with such laws and we conduct training sessions on these laws and codes, it is possible that enforcement authorities may view our relationships as prohibited arrangements that must be restructured or for which we would be subject to other significant civil or criminal penalties or debarment. In any event, any enforcement review of or action against us as a result of such review, regardless of outcome, could be costly and time consuming. Additionally, we cannot predict the impact of any changes in or interpretations of these laws, whether these changes will be retroactive or will have effect on a going-forward basis only.
Tax laws, regulations, and enforcement practices are evolving and may have a material adverse effect on our results of operations, cash flows and financial position.
Tax laws, regulations, and administrative practices in various jurisdictions are evolving and may be subject to significant changes due to economic, political, and other conditions. There are many transactions that occur during the ordinary course of business for which the ultimate tax determination is uncertain, and significant judgment is required in evaluating and estimating our provision and accruals for taxes. Additionally, the Australian Taxation Office’s interpretation of specific expenditures’ eligibility may vary, potentially leading to variances to our estimations. Governments are increasingly focused on ways to increase tax revenues, particularly from multinational corporations, which may lead to an increase in audit activity and aggressive positions taken by tax authorities.
Developments in relevant tax laws, regulations, administrative practices and enforcement practices could have a material adverse effect on our operating results, financial position and cash flows, including the need to obtain additional financing.
We are subject to tax audits by various tax authorities in many jurisdictions.
Our income tax returns are based on calculations and assumptions that require significant judgment and are subject to audit by various tax authorities. In addition, the calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws. We regularly assess the potential outcomes of examinations by tax authorities in determining the adequacy of our provision for income taxes.
Risks Related to Intellectual Property
Our success depends on our ability to protect our intellectual property and our proprietary technology.
Our success is to a certain degree also dependent on our ability to obtain and maintain patent protection. We could be materially adversely affected by any failure or inability to protect our intellectual property rights.
Similarly, any know-how that is proprietary or particular to our technologies could be subject to risk of disclosure by employees or consultants despite having confidentiality agreements in place.
Any future success will depend in part on whether we can obtain and maintain patents to protect our own products and technologies; obtain licenses to the patented technologies of third parties; and operate without infringing on the proprietary rights of third parties. Patent matters can involve complex legal and scientific questions and it is impossible to predict the outcome of patent claims. There is a risk that future patent applications that we make may not be approved, or we may not develop additional products or processes that are patentable. Some countries in which we may sell our products or license our intellectual property may fail to protect our intellectual property rights to the same extent as the protection that may be afforded in the United States or Australia.
In addition, the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Changes in either patent laws or in interpretations of patent laws could diminish the value of our intellectual property or narrow the scope of our patent protection. Even if we are able to obtain patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. We may also fail to take the required actions or pay the necessary fees to maintain our patents.
Moreover, any of our pending applications may be subject to a third-party pre-issuance submission of prior art to the United States Patent and Trademark Office (“USPTO”), the European Patent Office, the Intellectual Property Office in the United Kingdom, and the Australian Patent and Trademark Office. In addition, any patents issued could become involved in opposition, derivation, reexamination, post-grant review, interference proceedings or other patent office proceedings or litigation challenging our patent rights. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, and allow third parties to commercialize our technology or products and compete directly with us, without payment to us. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to exploit our intellectual property or develop or commercialize current or future products.
The issuance of a patent is not conclusive as to the inventorship, scope, validity or enforceability and our patents could be challenged in the courts or patent offices. Such challenges could result in loss of ownership or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit the duration of the patent protection of our technology and products. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, other companies may attempt to circumvent any regulatory data protection or market exclusivity that we obtain under applicable legislation and thus require us to allocate significant resources to preventing such circumvention. Such developments could enable other companies to circumvent our intellectual property rights and use our clinical trial data to obtain marketing authorizations in certain jurisdictions. Such developments could also require us to allocate significant resources to prevent other companies from circumventing or violating our intellectual property rights.
Intellectual property rights of third parties could adversely affect our ability to commercialize our products.
Our commercial success may depend upon our future ability and the ability of our potential collaborators to develop, manufacture, market and sell our products without infringing valid intellectual property rights of third parties. There is a substantial amount of litigation involving patents and other intellectual property rights in the medical technology industry, as well as administrative proceedings for challenging patents, including interference, derivation, inter partes review, post-grant review and reexamination proceedings before the USPTO or oppositions and other comparable proceedings in foreign jurisdictions. Furthermore, patent reform and changes to patent laws in the United States and in foreign jurisdictions add uncertainty to the possibility of challenge to our patents in the future, and could diminish the value of patents in general, thereby impairing our ability to protect our products. We cannot assure you that our products and other proprietary technologies we may develop will not infringe existing or future patents owned by third parties. Litigation or other legal proceedings relating to intellectual property claims, with or without merit, is unpredictable and generally expensive and time consuming and, even if resolved in our favor, is likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities.
If a third-party intellectual property right exists it could require the pursuit of litigation or administrative proceedings to nullify or invalidate the third-party intellectual property right concerned, or entry into a license agreement with the intellectual property right holder, which may not be available on commercially reasonable terms, if at all. Third-party intellectual property right holders, including our competitors, may bring infringement claims against us. If a third-party claims that we infringe its intellectual property rights, we may face a number of issues, including, but not limited to:
•
litigation, which may be expensive and time-consuming and may divert our management’s attention from our core business;
•
substantial damages for infringement, which we may have to pay if a court decides that the product or technology at issue infringes on or violates the third party’s rights, and, if the court finds that the infringement was willful, we could be ordered to pay treble damages and the patent owner’s attorneys’ fees;
•
a court prohibiting us from developing, manufacturing, marketing or selling our products, or from using our proprietary technologies, unless the third-party licenses its product rights to us, which it is not required to do;
•
if a license is available from a third party, we may have to pay substantial royalties, upfront fees and other amounts, and/or grant cross-licenses to intellectual property rights for our products; and
•
redesigning our products or processes so they do not infringe third-party intellectual property rights, which may not be possible or may require substantial monetary expenditures and time.
Numerous United States and foreign issued patents and pending patent applications owned by third parties exist in the fields in which we are developing our products. We cannot provide any assurances that valid third-party patents do not exist which might be enforced against our current or future products, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties. As the medical technology industry expands and more patents are issued, the risk increases that our products may give rise to claims of infringement of the patent rights of others. Third parties may assert that we infringe their patents or other intellectual property, or that we are otherwise employing their proprietary technology without authorization and may sue us. We believe that we have reasonable defenses against possible allegations of infringement, such as noninfringement or invalidity defenses; however, there can be no assurance that these defenses will succeed. It is also possible that patents owned by third parties of which we are aware or might become aware, but which we believe are not valid, or do not believe are relevant to our products and other proprietary technologies we may develop, could be found to be infringed by our products. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our products may infringe. In addition, our competitors or other third parties, many of which have substantially greater resources than we do and have made substantial investments in patent portfolios and competing technologies, may obtain patents in the future that may prevent, limit or otherwise interfere with our ability to make, use and sell our products, and may claim that use of our technologies or the manufacture, use or sale of our products infringes upon these patents. If any such third-party patents were held by a court of competent jurisdiction to cover our technologies or products, or if we are found to otherwise infringe a third party’s intellectual property rights, the holders of any such patents may be able to block, including by court order, our ability to develop, manufacture or commercialize the applicable product unless we obtain a license under the applicable patents or other intellectual property, or until such patents expire or are finally determined to be held invalid or unenforceable. Such a license may not be available on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. If we are unable to obtain a necessary license to a third-party patent on commercially reasonable terms, our ability to commercialize our products may be impaired or delayed, which could in turn significantly harm our business.
The medical technology industry has produced a considerable number of patents, and it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we were sued for patent infringement, we would need to demonstrate that our products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity may be difficult. For example, in the United States, proving invalidity in court requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents, and there is no assurance that a court of competent jurisdiction would invalidate the claims of any such United States patent. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on our business and operations. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.
Third parties asserting their patent or other intellectual property rights against us may seek and obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize our products or force us to cease some of our business operations. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of management and other employee resources from our business, cause development delays and may impact our reputation. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible on a cost-effective basis or require substantial time and monetary expenditure. In that event, we would be unable to further develop and commercialize our products, which could harm our business significantly. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we collaborate with various organizations and academic institutions on the advancement of our technology and products, we may, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite these contractual provisions, the need to share trade secrets and other confidential information increases the risk that such trade secrets will become known by potential competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, discovery by a third party of our trade secrets or other unauthorized use or disclosure would impair our intellectual property rights and protections in our products.
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In some cases, publication rights are controlled exclusively by us. In other cases, we may share these rights with other parties. Despite our efforts to protect our trade secrets, our competitors could discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the USPTO and other governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The USPTO and various corresponding governmental patent agencies outside of the United States require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
Confidentiality and invention assignment agreements with our employees, advisors and consultants may not adequately prevent disclosure of trade secrets and protect other proprietary information.
We consider proprietary trade secrets and/or confidential know-how and unpatented know-how to be important to our business. We may rely on trade secrets and/or confidential know-how to protect our technology, especially where patent protection is believed by us to be of limited value. However, trade secrets and/or confidential know-how can be difficult to maintain as confidential.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, advisors and consultants to enter into confidentiality and invention assignment agreements with us. However, current or former employees, advisors and consultants could unintentionally or willfully disclose our confidential information to competitors, and confidentiality and invention assignment agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party obtained illegally and is using trade secrets and/or confidential know-how is expensive, time consuming and unpredictable. The enforceability of confidentiality and invention assignment agreements may vary from jurisdiction to jurisdiction.
Failure to obtain or maintain trade secrets and/or confidential know-how trade protection could adversely affect our competitive position. Moreover, our competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors could limit our use of our trade secrets and/or confidential know-how.
Intellectual property rights do not address all potential threats to our business prospects.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
●
Others may be able to make products that are similar to ours but that are not covered by our intellectual property rights.
●
Others may independently develop similar or alternative technologies or otherwise circumvent any of our technologies without infringing our intellectual property rights.
●
We or any of our collaboration partners might not have been the first to conceive and reduce to practice the inventions covered by the patents or patent applications that we own, license or will own or license.
●
We or any of our collaboration partners might not have been the first to file patent applications covering certain of the patents or patent applications that we or they own or have obtained a license.
●
It is possible that any pending patent applications that we have filed, or will file, will not lead to issued patents.
●
Issued patents that we own may not provide us with any competitive advantage, or may be held invalid or unenforceable, as a result of legal challenges by our competitors.
●
Our competitors might conduct R&D activities in countries where we do not have patent rights, or in countries where R&D safe harbor laws exist, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets.
●
Ownership of our patents or patent applications may be challenged by third parties.
●
Our patents may only be valid for a limited period of time.
●
The patents of third parties or pending or future applications of third parties, if issued, may have an adverse effect on our business.
Any difficulty with protecting our intellectual property could diminish the value of our intellectual property rights in the relevant jurisdiction.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States, the United Kingdom, the European Union and Australia. If we or our collaboration partners encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in other jurisdictions, then the value of these rights could be diminished and we could face additional competition from others in such other jurisdictions.
Some countries in Europe and China have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we are, or any of our licensors is, forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position or commercial advantage may be impaired and our business and results of operations may be adversely affected.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our products and any future products.
The United States Supreme Court in recent years has issued rulings either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations or ruling that certain subject matter is not eligible for patent protection. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by Congress, the federal courts, the USPTO and equivalent bodies in non-United States jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce existing patents and patents we may obtain in the future.
Risks Related to Our Common Stock
Medtronic beneficially owns a significant equity interest in us and its interests may conflict with our or your interests.
As of January 22, 2026, as reported on the Schedule 13D filed by Medtronic on January 29, 2026, Medtronic beneficially owned approximately 16.1% of our total Common Stock. In light of such ownership, Medtronic is in a position to exercise influence over matters affecting our stockholders or requiring stockholder approval, including the election of the Board, amendments to our governing documents and the determination of significant corporate actions. Additionally, pursuant to the Investor Rights Agreement and Registration Rights Agreement, Medtronic has certain rights, and the ability to take certain actions, which are not otherwise available to all stockholders. For example, pursuant to the Investor Rights Agreement, Medtronic has certain rights, including participation rights with respect to future issuances of our equity securities, the right to designate one non-voting board observer to our Board, and the right to negotiate should we receive certain acquisition proposals.
Additionally, the Registration Rights Agreement provides Medtronic the right to demand that we file a registration statement to cover the resale by Medtronic of the shares of Common Stock sold to it in the Medtronic Private Placement. We have also granted Medtronic the right to demand the sale of these shares of Common Stock in an underwritten offering. For more information on the provisions of, including the specific conditions of the above-described rights granted in, the Investor Rights Agreement or Registration Rights Agreement, see “Item 13. Certain Relationships and Related Transactions, and Director Independence” of Part III of this Form 10-K.
The interests of Medtronic may not align with the interests of our other stockholders. Medtronic, as a global healthcare technology company, directly competes with us and may acquire and hold interests in businesses that compete directly or indirectly with us. Medtronic may also pursue acquisition opportunities that may be complementary to our business, and, as a result, those acquisition opportunities may not be available to us.
We will require substantial additional future financing and, until commercialization of our products, our cash burn in future periods may be higher than anticipated. We may be unable to raise sufficient capital in future financings, including to account for unanticipated cash burn, which could have a material impact on our R&D programs or commercialization of our products.
Developing medical device products, including conducting clinical trials and preclinical studies, is a very time-consuming, expensive and uncertain process that takes years to complete. While we have raised substantial funds through the capital markets in our initial public offering, 2026 Public Offering, 2025 Private Placement and Medtronic Private Placement, we will require additional financing in the future. Our operations have consumed substantial amounts of cash since inception, and our expenses will continue to increase in connection with our ongoing activities, particularly as we conduct our ongoing and planned preclinical studies and clinical trials of, and seek regulatory clearance and approval for, our current products, including DurAVR® THV System, and future products we may develop or otherwise acquire. Even if one or more of our products is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product, including manufacturing and supply costs, as well as costs associated with establishing a sales and end-to-end supply chain management infrastructure.
We have historically devoted most of our financial resources to R&D. To date, we have financed a significant amount of our operations through equity financings, including our initial public offering, and to a lesser extent, through the divestment of business segments and incurrence of indebtedness and convertible indebtedness. The amount of our future net losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity or debt financings or strategic collaborations. Our future capital requirements will depend on many factors, including but not limited to:
•
the scope, timing, progress, costs and results of discovery, preclinical development and clinical trials for our current or future products;
•
the number and size of clinical trials required for regulatory clearance and approval of our current or future products;
•
the costs, timing and outcome of regulatory review of any of our current or future products;
•
the costs associated with acquiring or licensing additional products, technologies or assets;
•
the cost of manufacturing clinical and commercial supplies of our current or future products;
•
the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending against any intellectual property-related claims, including any claims by third parties that we are infringing upon their intellectual property rights;
•
our ability to maintain existing, and establish new, strategic collaborations or other arrangements and the financial terms of any such agreements;
•
the costs and timing of future commercialization activities, including manufacturing, marketing, sales and end-to-end supply chain management, for any of our products for which we receive regulatory clearance and approval;
•
the revenue, if any, received from commercial sales of our products for which we receive regulatory clearance and approval;
•
expenses to attract, hire and retain skilled personnel;
•
the costs of operating as a dual-listed public company;
•
our ability to establish a commercially viable pricing structure and obtain approval for coverage and adequate reimbursement from third-party and government payors;
•
the effect of competing technological and market developments; and
•
the extent to which we acquire or invest in additional businesses, products and technologies.
The amount of such future net losses, as well as the possibility of future profitability, will also depend on our success in developing and commercializing products that generate significant revenue. Until our products become commercially available, we will need to obtain additional funding in connection with the further development of our products. Our ability to obtain additional financing will be subject to a number of factors, including market conditions, our operating performance and investor sentiment. As such, additional financing may not be available to us when needed, on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our R&D programs or any future commercialization efforts or obtain funds by entering agreements on unattractive terms.
Furthermore, any additional equity and equity-linked fundraising in the capital markets may be dilutive for stockholders and any debt-based funding may bind us to restrictive covenants and curb our operating activities and ability to pay potential future dividends even when profitable. In addition, the issuance of additional equity and equity-linked securities by us, or the possibility of such issuance, may cause the market price of our Common Stock to decline. We cannot guarantee that future financing will be available in sufficient amounts or on acceptable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on acceptable terms, we will be prevented from pursuing R&D efforts. This could harm our business, operating results and financial condition and cause the price of our Common Stock to fall.
Our Common Stock is listed on Nasdaq, but the market price and trading volume of our Common Stock may be volatile and may be affected by economic conditions beyond our control.
The market price of our Common Stock may be highly volatile and subject to wide fluctuations. In addition, the trading volume of our Common Stock may fluctuate and cause significant price variations to occur. If the market price of our Common Stock declines significantly, you may be unable to resell your Common Stock at a competitive price. We cannot assure you that the market price of our Common Stock will not fluctuate or significantly decline in the future.
Some specific factors that could negatively affect the price of our Common Stock or result in fluctuations in their price and trading volume include:
•
actual or expected fluctuations in our prospects or operating results;
•
announcements relating to our products, including the results of clinical trials by us or our collaborators;
•
changes in the demand for our products;
•
additions or departures of our key personnel;
•
changes or proposed changes in laws, regulations or tax policy;
•
sales or perceived potential sales of our Common Stock by us or our executive officers, directors or stockholders in the future;
•
announcements or expectations concerning additional financing efforts; and
•
conditions in the United States, Australian and global financial markets, or in our industry in particular, or changes in general economic or political conditions.
In recent years, the stock market in general, and the market for medical technology companies in particular, has experienced significant price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Broad market and industry factors may seriously affect the market price of our Common Stock, regardless of our actual operating performance.
When the market price of a stock has been volatile, as our Common Stock price may be, holders of that stock have occasionally brought securities class action litigation claims against the company that issued the stock. If any of our stockholders were to bring a lawsuit of this type against us, even if the lawsuit were without merit, we could incur substantial costs defending the lawsuit. The lawsuit could also divert the time and attention of our management.
Shares of our Common Stock are listed to trade on Nasdaq in United States dollars and our CDIs are listed to trade on the ASX in Australian dollars, which may result in price variations.
Shares of our Common Stock are listed to trade on Nasdaq in United States dollars and our CDIs are listed to trade on the ASX in Australian dollars. Dual-listing may result in price variations between the exchanges due to a number of factors. In addition, the exchanges are open for trade at different times of the day and the two exchanges also have differing vacation schedules. Differences in the trading schedules, as well as volatility in the exchange rate of the two currencies, among other factors, may result in different trading prices for our Common Stock and CDIs on the two exchanges. Other external influences may have different effects on the trading price of our Common Stock and CDIs on the two exchanges.
An active, liquid trading market for our Common Stock may not be maintained.
We can provide no assurance that we will be able to maintain an active trading market for our Common Stock. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. An inactive market may also impair our ability to raise capital by selling our Common Stock and our ability to acquire other companies, products or technologies by using our Common Stock as consideration.
We do not anticipate paying dividends in the foreseeable future.
ATPL (which became a subsidiary of the Company following completion of the Reorganization) did not declare any dividends during fiscal years 2022, 2023, 2024 or 2025 and we do not anticipate that we will do so in the foreseeable future. We currently intend to retain future earnings, if any, to finance the development of our business. Dividends, if any, on our outstanding Common Stock will be declared by and subject to the discretion of our Board on the basis of our earnings, financial requirements and other relevant factors, and subject to Delaware and federal law. We cannot assure you that our Common Stock will appreciate in value. You may not realize a return on your investment in our Common Stock and you may even lose your entire investment in our Common Stock.
If United States securities or industry analysts do not publish research reports about our business, or if they issue an adverse opinion about our business, the market price and trading volume of our Common Stock could decline.
The trading market for our Common Stock will be influenced by the research and reports that United States securities or industry analysts publish about us or our business. Securities and industry analysts may discontinue research on us, to the extent such coverage currently exists, or in other cases, may never publish research on us. If no or too few United States securities or industry analysts commence coverage of our company, the trading price for our Common Stock would likely be negatively affected. In the event securities or industry analysts initiate coverage, if one or more of the analysts who cover us downgrade our Common Stock or publish inaccurate or unfavorable research about our business, the market price of our Common Stock would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our Common Stock could decrease, which might cause our price and trading volume to decline. In addition, research and reports that Australian securities or industry analysts may, initiate or may continue to, publish about us, our business or our Common Stock may impact the market price of our Common Stock.
We are an “emerging growth company” and a “smaller reporting company” and our election of reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our Common Stock less attractive to investors and, as a result, adversely affect the price of our Common Stock and result in a less active trading market for our Common Stock.
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”) and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. For example, we have elected to rely on an exemption from the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) relating to internal control over financial reporting (“ICFR”), reduced disclosure obligations regarding executive compensation in this Form 10-K and our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation. In addition, as a smaller reporting company, we are only required to provide two years of audited financial statements.
We may avail ourselves of these disclosure exemptions until we are no longer an “emerging growth company.” We cannot predict whether investors will find our Common Stock less attractive because of our reliance on some or all of these exemptions. If investors find our Common Stock less attractive, it may adversely affect the price of our Common Stock and there may be a less active trading market for our Common Stock.
We will cease to be an “emerging growth company” upon the earliest of:
•
the last day of the fiscal year during which we have total annual gross revenues of $1,235,000,000 (as such amount is indexed for inflation every five years by the SEC) or more;
•
the last day of our fiscal year following the fifth anniversary of the closing of our initial public offering;
•
the date on which we have, during the previous three-year period, issued more than $1,000,000,000 in non-convertible debt; or
•
the date on which we are deemed to be a “large accelerated filer,” as defined in Rule 12b-2 of the Exchange Act.
Furthermore, Section 102(b)(1) of the JOBS Act exempts emerging growth companies from being required to comply with new or revised financial accounting standards until private companies (that is, those that have not had a Securities Act registration statement declared effective or do not have a class of securities registered under the Exchange Act) are required to comply with the new or revised financial accounting standards. The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies but such an election to opt out is irrevocable. We have elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public or private companies, we, as an emerging growth company, can adopt the new or revised standard at the time private companies adopt the new or revised standard, until such time we are no longer considered to be an emerging growth company. This may make comparison of our financial statements with another public company which is neither an emerging growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible because of the potential differences in accounting standards used.
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies for so long as we are a smaller reporting company.
We incur increased costs as a result of operating as a United States listed public company, and our management is required to devote substantial time to new compliance initiatives and corporate governance practices, which could divert their attention from the operation of our business.
As a United States listed public company, and particularly after we are no longer an “emerging growth company,” we incur, and will continue to incur, significant additional legal, accounting, and other expenses. The Dodd-Frank Wall Street Reform and Consumer Protection Act (“Dodd-Frank Act”), the Sarbanes-Oxley Act, the listing requirements of Nasdaq, and other applicable securities rules and regulations impose various requirements on public companies, including the filing of reports with respect to our business and operating results, establishment and maintenance of effective disclosure controls and procedures, maintenance and reporting of our system of ICFR, and other corporate governance practices. We expect that we will need to hire additional accounting, finance, legal, and other personnel in connection with our efforts to comply with the requirements of being a United States public company, and our management and other personnel need to devote a substantial amount of time towards maintaining compliance with these requirements. These requirements increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
We have identified material weaknesses in our internal control over financial reporting. If we fail to remediate these material weaknesses, or if we experience additional material weaknesses in the future or otherwise fail to maintain effective internal control over financial reporting in the future, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in us and, as a result, the value of our Common Stock.
In connection with the preparation of our financial statements for the years ended December 31, 2025 and 2024, our management and our independent auditors identified material weaknesses in the design and operating effectiveness of our ICFR. Additionally, as a public company, we are required under Section 404 of the Sarbanes‑Oxley Act to provide an annual report by management on the effectiveness of our ICFR. In connection with our evaluation as of December 31, 2025, management concluded that our ICFR was not effective due to these material weaknesses, which had not yet been fully remediated. A material weakness is a deficiency, or combination of deficiencies, in ICFR, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis.
The material weaknesses identified by our management and our independent auditors relate to (i) a lack of appropriately designed, implemented and documented procedures and controls, and (ii) deficiencies in the segregation of duties.
To remediate these material weaknesses, management has initiated a remediation plan, which includes formally documenting policies, processes, risks, and controls, and reviewing the design and operating effectiveness of key and non-key controls. Segregation of duties has been enhanced across the control environment and financial reporting systems through system automation, strengthened month-end controls, and strengthened month-end review procedures. Testing of operating effectiveness has commenced. Remediation actions are progressing as planned.
While we believe that these efforts will improve our ICFR, the design and implementation of our remediation is ongoing and will require validation and testing of the design and operating effectiveness of our internal controls over a sustained period of financial reporting cycles. The actions that we are taking are subject to ongoing senior management review, as well as oversight by our Audit and Risk Committee. We will not be able to conclude whether the steps we are taking will fully remediate the material weaknesses in our ICFR until we have completed our remediation efforts and subsequent evaluation of their effectiveness.
When we cease to be an “emerging growth company” under the federal securities laws, our independent registered public accounting firm may be required to express an opinion on the effectiveness of our ICFR. If we are unable to confirm that our ICFR is effective, or if our independent registered public accounting firm is unable to express an unqualified opinion on the effectiveness of our ICFR, we could lose investor confidence in the accuracy and completeness of our financial reports, which could cause the price of our Common Stock to decline. We could also become subject to investigations by Nasdaq, the SEC or other regulatory authorities, and become subject to litigation from investors and stockholders, which could harm our reputation and financial condition or divert financial and management resources from our regular business activities
Our Charter and Bylaws contain anti-takeover provisions that could delay or discourage takeover attempts that stockholders may consider favorable.
Our Charter and Bylaws contain provisions that could delay or prevent a change in control of our company. These provisions could also make it difficult for stockholders to elect directors who are not nominated by the current members of our Board or take other corporate actions, including effecting changes in our management. These provisions include:
•
the ability of our Board to issue shares of preferred stock and to determine the price and other terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer;
•
a staggered Board divided into three classes serving staggered three-year terms, such that not all members of our Board will be elected at one time;
•
allowing only our Board to fill director vacancies, which prevents stockholders from being able to fill vacancies on our Board;
•
a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders;
•
a requirement for the affirmative vote of holders of at least 75% of the voting power of all of the then-outstanding shares of the voting stock, voting together as a single class, to amend certain provisions of our Charter or our Bylaws, which may inhibit the ability of an acquirer to effect such amendments to facilitate an unsolicited takeover attempt;
•
the ability of our Board to amend our Bylaws, which may allow our Board to take additional actions to prevent an unsolicited takeover and inhibit the ability of an acquirer to amend the Bylaws to facilitate an unsolicited takeover attempt;
•
advance notice procedures with which stockholders must comply to nominate candidates to our Board or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of our company; and
•
a prohibition of cumulative voting in the election of our Board, which would otherwise allow less than a majority of stockholders to elect director candidates.
Future equity financings and sales by existing holders could adversely affect the voting power or value of our Common Stock.
We may from time to time raise funds through the issuance of Common Stock or the issuance of debt instruments or other securities convertible into Common Stock. We cannot predict the size or price of future issuances of Common Stock or the size or terms of future issuances of debt instruments or other securities convertible into Common Stock, or the effect, if any, that future issuances and sales of our securities will have on the market price of the Common Stock. Sales or issuances of substantial numbers of shares of Common Stock, or the perception that such sales or issuances could occur, may adversely affect prevailing market prices of the Common Stock. With any additional sale or issuance of Common Stock, or securities convertible into Common Stock, investors will suffer dilution to their voting power and we may experience dilution in our earnings per share.
Our Charter authorizes us to issue, without the approval of our stockholders, one or more classes or series of preferred stock having such designations, preferences, limitations and relative rights, including preferences over our Common Stock respecting dividends and distributions, as our Board may determine. The terms of one or more classes or series of preferred stock could adversely impact the voting power or value of our Common Stock. For example, we might grant holders of preferred stock the right to elect some number of our directors in all events or on the happening of specified events or the right to veto specified transactions. Similarly, the repurchase or redemption rights or liquidation preferences we might grant to holders of preferred stock could affect the residual value of our Common Stock.
Our failure to meet Nasdaq’s continued listing requirements could result in a delisting of our Common Stock.
If we fail to satisfy the continued listing requirements of Nasdaq, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our Common Stock. Such a delisting would likely have a negative effect on the price of our Common Stock and would impair your ability to sell or purchase our Common Stock when you wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow our Common Stock to become listed again, stabilize the market price or improve the liquidity of our Common Stock, prevent our Common Stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with the listing requirements of Nasdaq.
If Nasdaq delists our Common Stock from trading on its exchange, we could face significant material adverse consequences, including:
•
a limited availability of market quotations for our Common Stock;
•
a determination that our Common Stock is a “penny stock” which will require brokers trading in our Common Stock to adhere to more stringent rules, which could result in a reduced level of trading activity in the secondary trading market for our Common Stock;
•
more limited news and analyst coverage for our company; and
•
a decreased ability to issue additional securities or obtain additional financing in the future.
We are a holding company and, as such, we will depend on our subsidiaries to support our operations.
We are a holding company and essentially all of our assets are the capital stock of our subsidiaries. As a result, investors in our company are subject to the risks attributable to our subsidiaries. As a holding company, we conduct all of our business through our subsidiaries. Therefore, our ability to fund and conduct our business, service our debt and pay dividends, if any, in the future will principally depend on the ability of our subsidiaries to continue their R&D activities and, post-commercialization, generate sufficient cash flow to make upstream cash distributions to us. Our subsidiaries are separate legal entities, and although they are controlled by us, they have no obligation to make any funds available to us, whether in the form of loans, dividends or otherwise. The ability of these entities to pay dividends and other distributions will depend on their operating results and will be subject to applicable laws and regulations which require that solvency and capital standards be maintained by such companies and contractual restrictions contained in the instruments governing any debt obligations. In the event of bankruptcy, liquidation or reorganization of any of our material subsidiaries, holders of indebtedness and trade creditors may be entitled to payment of their claims from the assets of those subsidiaries before our company.
Our Bylaws designate the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or agents.
Our Bylaws provide that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware (or, if the Court of Chancery does not have jurisdiction, the federal district court for the District of Delaware) will, to the fullest extent permitted by applicable law, be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers, employees or agents to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the DGCL, our Charter or Bylaws or (iv) any action asserting a claim against us that is governed by the internal affairs doctrine, in each such case subject to such Court of Chancery of the State of Delaware having personal jurisdiction over the indispensable parties named as defendants therein. Our Bylaws further provide that, unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States will, to the fullest extent permitted by law, be the sole and exclusive forum for the resolutions of any complaint asserting a cause of action arising under the Securities Act. We note that there is uncertainty as to whether a court would enforce the choice of forum provision with respect to claims under the Securities Act, and that investors cannot waive compliance with the Securities Act and the rules and regulations thereunder. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock will be deemed to have notice of, and consented to, the provisions of our Bylaws described in the preceding sentence. This forum selection provision is not intended to apply to any actions brought under the Exchange Act. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder.
These choice-of-forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers, employees or agents, which may discourage such lawsuits against us and such persons. These choice-of-forum provisions may also impose additional litigation costs on stockholders in pursuing any such claims against us and/or our directors, officers, employees, or agents, to the extent the provisions require the stockholders to litigate in a particular or different forum. In addition, while the Delaware Supreme Court ruled in March 2020 that federal forum selection provisions purporting to require claims under the Securities Act be brought in federal court are “facially valid” under Delaware law, there is uncertainty as to whether other courts will enforce our choice-of-forum provisions. Our choice-of-forum provisions may impose additional litigation costs on stockholders who assert that the provisions are not enforceable or invalid.
Alternatively, if a court were to find these provisions of our Bylaws inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, financial condition or operating results.
Our ability to use our United States net operating loss carryforwards to offset future taxable income may be subject to certain limitations.
As of December 31, 2025 we had United States federal net operating loss (“NOL”) carryforwards of $158.8 million, which may be available to offset federal income tax liabilities in the future. In addition, we may generate additional NOLs in future years. In general, a corporation’s ability to utilize its NOLs may be limited if it experiences an “ownership change” as defined in Section 382 of the United States Internal Revenue Code of 1986, as amended (the “Code”). An ownership change generally occurs if certain direct or indirect “5-percent shareholders,” as defined in Section 382 of the Code, increase their aggregate percentage ownership of a corporation’s stock by more than 50 percentage points over their lowest percentage ownership at any time during the testing period, which is generally the three-year period preceding any potential ownership change. If a corporation experiences an ownership change, the corporation will be subject to an annual limitation that applies to the amount of pre-ownership change NOLs that may be used to offset post-ownership change taxable income. This limitation is generally determined by multiplying the value of the corporation’s stock immediately before the ownership change by the applicable long-term tax-exempt rate. Any unused annual limitation may, subject to certain limits, be carried over to later years, and the limitation may under certain circumstances be increased by built-in gains in the assets held by such corporation at the time of the ownership change. Similar rules and limitations may apply for state income tax purposes.
Our initial public offering in the United States in December 2024 caused an “ownership change" for Anteris Technologies Corporation. Under this “ownership change,” Section 382 of the Code will impose an annual limit on the amount of pre-ownership change NOL carryforwards and other tax attributes we could use to reduce our taxable income, potentially increasing and accelerating our liability for income taxes. This “ownership change” is not expected to cause any tax attributes to expire unused. It is possible that any future ownership changes could materially reduce our ability to use our United States NOL carryforwards or other tax attributes to offset taxable income, which could adversely affect our profitability.
No further ownership changes during the year to December 31, 2025 were noted in the United States. Following the capital raises in January 2026, further examination of loss carryforwards is being performed. A potential change in ownership may subject loss carryforwards in our U.S. parent company to a limitation under Section 382 of the Internal Revenue Code. U.S. federal and state losses attributable to this entity are $25.6 million.
Our ability to use our Australian net operating and capital loss carryforwards to offset future taxable income are subject to the satisfaction of loss tests.
As of December 31, 2025, we had Australian net operating and capital loss (“NOCL”) carryforwards of $74.5 million, which may be available to offset Australian income tax liabilities in the future. In addition, we may generate additional NOCLs in future years.
In general, a corporation’s ability to utilize its NOCLs is impacted if it does not satisfy one of two loss tests - the continuity of ownership test (where there is a change in majority ownership and control) or failing that, the business continuity test. These tests are set forth in Divisions 165 and 166 of the Income Tax Assessment Act 1997. The loss tests are applied to each parcel of NOCLs that arise in a particular income year.
The continuity of ownership test is failed where a majority interest in shareholders’ rights to dividends, rights to capital distributions and voting rights are not maintained (i.e., there is a change in majority ownership and control). A concession applies whereby all shareholders with less than 10% of these rights are deemed to be held by a single notional shareholder. If the total of the single notional shareholder interests falls below 50%, the continuity of ownership test may be failed and the NOCLs (either all or particular parcels) may only be utilized if the business continuity test is satisfied.
The business continuity test considers whether ATPL has maintained a similar or same business at the relevant testing times.
Our initial public offering in the United States in December 2024 did not result in a failure of the continuity of ownership test. In addition, capital raises completed during 2025 and January 2026 also did not result in a failure of the continuity of ownership test. However, if future issuances or sales of our Common Stock, including certain transactions involving our Common Stock that are outside of our control, were to result in a failure of the continuity of ownership test and the business continuity test could not be satisfied, our NOCLs may be limited or unavailable to offset taxable income. As a result, our income tax liabilities could increase and be accelerated.
Sales of a substantial number of shares of our Common Stock in the public market by our existing stockholders could cause the price of our Common Stock to fall.
The market price of shares of our Common Stock could decline as a result of sales of our Common Stock or CDIs representing those shares, including substantial sales by our significant stockholders, or the perception that these sales could occur. In addition, the future registration of shares of our Common Stock may cause our stock price to decline, even before such shares are actually sold in the market. We have registered shares of Common Stock that we may issue under our employee equity incentive plans. These shares can be sold freely in the public market upon issuance.
Further, we are party to a Registration Rights Agreement with Medtronic, which requires us to effect the registration of the shares of Common Stock sold to Medtronic in the Medtronic Private Placement for resale, and grant Medtronic the right to demand the sale of these shares of Common Stock in an underwritten offering. As such, sales of a substantial number of shares of our Common Stock in the public market could occur at any time. We are unable to predict the effect that sales, or the perception that our shares may be available for sale, will have on the prevailing market price of our Common Stock.
None.
Cybersecurity Risk Management and Strategy
The operation of our business depends on our information technology systems. We rely on our information technology systems to effectively manage sales and marketing data, accounting and financial functions, inventory management, product development tasks, clinical data, customer service and technical support functions. Our information technology infrastructure and products are vulnerable to cyber-based attacks. Cyber-based attacks can include computer viruses, denial-of-service attacks, phishing attacks, ransomware attacks and other introduction of malware to computers and networks; unauthorized access through the use of compromised credentials; exploitation of design flaws, bugs or security vulnerabilities; intentional or unintentional acts by employees or other insiders with access privileges; and intentional acts of vandalism by third parties and sabotage.
We have developed and implemented a risk-based program focused on cybersecurity measures to protect the confidentiality, integrity, and availability of our critical systems and information.
In addition to the annual security assessment and penetration test, the SGTM utilizes an Endpoint Detection and Response system which acts as an anti-virus and intrusion prevention system and reports all abnormalities to the SGTM. Essential Eight and Center for Internet Security Control Strategies, including patch applications, multi-factor authentication, administrative privileges, application hardening and backups, are performed internally via a security consultant with regular updates provided to the SGTM. Reported findings and action items are recorded and assigned to personnel for action.
Any failure to protect our information technology infrastructure and our products against cyber-based attacks, network security breaches, service interruptions or data corruption could materially disrupt our operations and harm our business.
Our cybersecurity risk management program includes:
•
periodic risk assessments;
•
annual security assessment and penetration testing;
•
•
the use of external service providers, where appropriate, to assess, test or otherwise assist with aspects of our security controls;
•
•
cybersecurity awareness training and phishing campaigns for specific employee groups;
•
disaster recovery plans/procedures;
•
access control and CCTV systems (where appropriate) for the physical protection of Anteris systems; and
•
incident response and recovery procedures, for cybersecurity incidents.
The locations and uses of our material properties are as follows:
|
Location of Facility
|
Lease expiry date | Extension options | |
|
11600-11628 96th Avenue North, Maple Grove, MN 55369 ("610 Maple Grove")(1)
|
April 30, 2030 | 1 period of two years | |
|
26 Harris Road, Malaga WA 6090, Australia
|
July 31, 2026 | - |
(1) Predominantly used for R&D, manufacturing of the DurAVR® valve and regulatory compliance teams. On April 30, 2025, we cancelled our sublease of the building without incurring a termination fee and entered into a new 5-year lease agreement directly with the landlord. As a result, we recognized a right-of-use (“ROU”) asset and a corresponding lease liability of $1.6 million on the lease commencement date. This change provides longer-term access to the premises.
All properties are leased or subleased. Our properties are well maintained, are in good operating condition, and are suitable for current requirements. We do not anticipate difficulty in renewing existing leases as they expire or in finding alternative facilities.
In the ordinary course of our operations, and from time-to-time, we are party to various claims and lawsuits. We are not party to any material legal proceedings, and no such proceedings are, to management’s knowledge, threatened against us.
Not applicable.
Item 5
Market Information
The Common Stock is traded on Nasdaq under the symbol “AVR.” The CDIs are traded on the ASX under the symbol “AVR.”
Holders of Record
As of December 31, 2025, there were 28 holders of record of the Common Stock on Nasdaq. This number includes a single holder of record for all shares of Common Stock underlying our CDIs.
The actual number of stockholders will be considerably greater than the number of stockholders of record and will include stockholders who are beneficial owners but whose shares of Common Stock are held in street name by brokers and other nominees.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock and we do not anticipate paying any cash dividends in the foreseeable future. We currently anticipate that we will retain all available funds for use in the operation and expansion of our business. Any future determination as to the declaration or payment of dividends on our Common Stock will be made at the discretion of our Board and will depend upon, among other factors, our financial condition, results from operations, current and anticipated cash needs, plans for expansion and other factors that our Board may deem relevant.
Equity Compensation Plan Information
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters of Part III of this Form 10-K.
Use of Proceeds from our Initial Public Offering
On December 12, 2024, the Registration Statement relating to our initial public offering became effective pursuant to which we issued and sold 14,878,481 shares of Common Stock at a public offering price of $6.00 per share. We received net proceeds of $80.0 million, after deducting the underwriting discounts, commissions and offering expenses and giving effect to the exercise of the underwriters’ option to purchase additional shares. None of the expenses associated with the initial public offering were paid to directors, officers, persons owning 10% or more of any class of equity securities, or to our affiliates.
The proceeds from the initial public offering were fully utilized in the manner described in the prospectus included in the Registration Statement.
Recent Sales of Unregistered Securities
None.
Issuer Repurchases of Equity Securities
During the quarter ended December 31, 2025, we did not repurchase any equity securities.
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) summarizes the significant factors affecting the operating results, financial condition and liquidity, and cash flows of our company for the year ended December 31, 2025. The Company was incorporated under the laws of the state of Delaware to become the holding company of our business pursuant to the Reorganization. Prior to completion of the Reorganization, the Company had no business or operations and, following completion of the Reorganization, the business and operations of the Company consists solely of the business and operations of ATGC and its subsidiaries. Our financial statements as of December 31, 2023 and as of and for the years ended December 31, 2024 and 2025 consolidate, and our future financial statements will consolidate ATGC as an operating subsidiary. This MD&A should be read in conjunction with our consolidated financial statements, the accompanying notes to consolidated financial statements and other financial information included in this Form 10-K. Except for historical information, the matters discussed in this MD&A contain various forward-looking statements that involve risks and uncertainties and are based upon judgments concerning various factors beyond our control. Our actual results could differ materially from those anticipated in these forward-looking statements. You should carefully read the section titled “Risk Factors” to gain an understanding of the important factors that could cause actual results to differ materially from our forward-looking statements. Please also see the section of this Form 10-K titled “Special Note Regarding Forward-Looking Statements”.
Overview
Anteris is a structural heart company dedicated to revolutionizing cardiac care by pioneering science-driven and measurable advancements to restore heart valve patients to healthy function. Our lead product, the DurAVR® THV System, was designed in collaboration with the world’s leading interventional cardiologists and cardiac surgeons to treat aortic stenosis — a potentially life-threatening condition resulting from a narrowing of the aortic valve. The balloon-expandable DurAVR® THV is the first biomimetic valve, which is shaped to mimic the performance of a healthy human aortic valve and aims to replicate normal aortic blood flow. Our DurAVR® THV System consists of a single-piece, biomimetic valve made with our proprietary ADAPT® tissue-enhancing technology and deployed with our balloon expandable ComASUR® Delivery System. ADAPT® is our proprietary anti-calcification tissue shaping technology that is designed to reengineer xenograft tissue into a pure, single-piece collagen bioscaffold. Our patented ADAPT® tissue has been clinically demonstrated to be calcium free for up to 10 years post-procedure, according to Performance of the ADAPT-Treated CardioCel® Scaffold in Pediatric Patients With Congenital Cardiac Anomalies: Medium to Long-Term Outcomes, published by William Neethling et. al., and has been distributed for use in over 55,000 patients globally in other indications. Our balloon expandable ComASUR® Delivery System, which was developed in consultation with physicians, is designed to provide precise alignment with the heart’s native commissures to achieve accurate placement of the DurAVR® THV. As of December 2025, more than 130 patients have been implanted with the DurAVR® THV worldwide.
In 2025, we advanced regulatory activities in Europe, with the goal of securing approval to commence the PARADIGM Trial in a number of European countries. In October 2025, we secured the first European regulatory approval in Denmark and subsequently enrolled and treated the first patients marking the formal initiation of the PARADIGM Trial. In November 2025, we also received IDE approval from the FDA for the PARADIGM Trial. The FDA granted a staged approval authorizing enrollment of the first 200 patients. We may request authorization to expand enrollment for the remaining subjects through an IDE supplement. Throughout the year, our cross‑functional teams continued to execute site activation, regulatory preparation, and operational readiness activities in anticipation of regulatory approval in each participating country.
We are a development stage company and have incurred net losses each year since operation, however, we believe that we have significant growth potential in a large, underpenetrated and growing TAVR market.
Financial Overview
As a development-stage company, we have incurred losses since our inception. We anticipate that we will continue to incur losses for the foreseeable future and there can be no assurance that we will ever achieve or maintain profitability.
We expect expenses for our research, clinical validation, development, design, manufacturing and marketing will increase and, as a result, we will need additional capital to fund our operations. Any future funding could involve a combination of equity offerings, debt financings, other third-party funding, marketing and distribution arrangements, strategic alliances and licensing arrangements. We may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms or at all.
Any failure to raise capital or enter into such other arrangements as and when needed could have a negative impact on our financial condition and our ability to market our products.
In October and November 2025, we completed the 2025 Private Placement, which generated gross proceeds totaling approximately $25.2 million.
In January 2026, we completed the 2026 Public Offering and the Medtronic Private Placement, which collectively generated gross proceeds of approximately $320 million, before deducting underwriting discounts and commissions, placement agent fees, and estimated offering expenses.
Principles of Consolidation and Operating Segments
The consolidated financial statements include the accounts for our company, our wholly-owned subsidiaries, and entities for which we have a controlling financial interest, and for periods prior to the Reorganization, the accounts of ATPL, its wholly-owned subsidiaries, and entities for which ATPL has a controlling financial interest. Intercompany transactions, balances and unrealized gains and losses on transactions between such entities are eliminated.
Our management has determined that the activities of the business as reviewed by the Vice Chairman and Chief Executive Officer, the chief operating decision maker (“CODM”), are one segment, being the development and commercialization of the ADAPT® anti-calcification tissue. This is focused on the DurAVR® THV System.
Components of Results of Operations
Revenue and Other Income
We derive revenue from the sale of regenerative tissue products. Such sales have historically been made principally to 4C and to LeMaitre Vascular, Inc. (“LeMaitre”), a distributor of medical products, to whom we sold the distribution rights for CardioCel™ and VascuCel™ in 2019 in order to focus on development of our proprietary ADAPT® tissue for the DurAVR® THV System. Concurrent with such sales, we entered into a transition services agreement (the “Transition Services Agreement”) with LeMaitre, pursuant to which we manufactured and sold CardioCel™ and VascuCel™ products to LeMaitre. The Transition Services Agreement with LeMaitre expired in January 2025. We recognized revenue from LeMaitre during January 2025 in accordance with the terms of the Transition Services Agreement; however, we do not expect to receive any ongoing revenues from LeMaitre associated therewith. We were also party to the 4C Agreement, a supply and license agreement with 4C, which had an initial seven-year term that expired on June 1, 2025, and under its terms would automatically renew for successive one-year periods unless either party provided written notice of non-renewal at least 180 days prior to the applicable renewal date. On November 26, 2025, we notified 4C that we were not renewing the 4C Agreement, which will terminate on June 1, 2026. We will not incur any early termination penalties in connection with its non-renewal of the 4C Agreement.
Expenses
Our most significant expenses are R&D and selling, general and administrative expenses.
Cost of products sold reflects the manufacturing cost from the sale of regenerative tissue products to 4C and to LeMaitre. These expenditures include raw materials and consumables, plus other costs attributable to the manufacturing of these products.
R&D Expense
R&D has been a significant focus for us with investments in the DurAVR® THV System, including the DurAVR® THV, the ComASUR® Delivery System, a disposable crimper, and an expandable access sheath, as we aim for commercialization. These components are collectively managed as part of the overall DurAVR® THV System rather than as separate projects. Since late 2021, when our DurAVR® THV was first used in human trials in Tbilisi, Georgia, R&D efforts have focused on incorporating feedback from the clinical trial and progressing towards commercialization. These costs have included, among others, preclinical and clinical studies, design iterations, laboratory services, clinical data monitoring, project and site management, travel, data management and safety of the study.
During 2025, we continued to expand global manufacturing capacity to scale for the PARADIGM Trial. All production has been, and will continue to be, scaled into new ISO Qualified Clean Room facilities, increasing manufacturing capacity relative to 2024 capacity levels. The transition to the new facilities aims for a reliable and scaled inventory supply to support the commencement of the PARADIGM Trial. In addition, the gold-standard ADAPT® tissue for the DurAVR® THV will be sourced from both the United States and Australia moving forward to help mitigate supply chain risks.
Results of Operations
Comparison of Years Ended December 31, 2025 and December 31, 2024
The following tables set forth our results of operations for the years ended December 31, 2025 and December 31, 2024 (in thousands, except percentages).
| Year Ended December 31, | ||||||||||||
| 2025 | 2024 | % Change | ||||||||||
|
Net sales
|
$ | 1,913 | $ | 2,703 | (29 | )% | ||||||
|
Costs and expenses:
|
||||||||||||
|
Cost of products sold
|
(569 | ) | (1,437 | ) | (60 | )% | ||||||
|
Research and development expense
|
(69,120 | ) | (51,451 | ) | 34 | % | ||||||
|
Selling, general and administrative expense
|
(26,118 | ) | (28,187 | ) | (7 | )% | ||||||
|
Operating loss
|
(93,894 | ) | (78,372 | ) | 20 | % | ||||||
|
Other non-operating income, net
|
467 | 2,442 | (81 | )% | ||||||||
|
Interest and amortization of debt discount and expense
|
(73 | ) | (47 | ) | 55 | % | ||||||
|
Net foreign exchange (losses)/gains
|
(744 | ) | 1,440 | (152 | )% | |||||||
|
Debt issuance costs
|
- | (465 | ) | (100 | )% | |||||||
|
Loss on debt extinguishment
|
- | (904 | ) | (100 | )% | |||||||
|
Fair value movement of derivatives
|
19 | (61 | ) | (131 | )% | |||||||
|
Loss before income taxes from continuing operations
|
(94,225 | ) | (75,967 | ) | 24 | % | ||||||
|
Income tax (expense)/benefit
|
- | - | - | |||||||||
|
Loss after income tax
|
(94,225 | ) | (75,967 | ) | 24 | % | ||||||
|
Total (loss)/gain is attributable to:
|
||||||||||||
|
Non-controlling interests
|
(81 | ) | 324 | (125 | )% | |||||||
|
Stockholders of the Company
|
$ | (94,144 | ) | $ | (76,291 | ) | 23 | % | ||||
Net Sales
Net sales in 2025 was $1.9 million, a decrease of $0.8 million (29%), compared to $2.7 million in 2024, primarily due to a decrease in sales of CardioCel™ and VascuCel™ products pursuant to the expiration of the LeMaitre Transition Services Agreement in January 2025, partly offset by increased demand for other higher-yielding tissue products in 2025.
Cost of Products Sold
Cost of products sold in 2025 was $0.6 million, a decrease of $0.9 million (60%), compared to $1.4 million in 2024, primarily due to a decrease in sales of CardioCel™ and VascuCel™ products following the expiration of the LeMaitre Transition Services Agreement in January 2025, partly offset by increased demand for other higher-yielding tissue products in 2025.
R&D Expense
R&D expenses in 2025 were $69.1 million, an increase of $17.7 million (34%) compared to $51.5 million in 2024. This is primarily due to an increase of $19.8 million related to the upscaling of manufacturing and quality capabilities, including process design and validation activities, and an increase in R&D headcount, an increase of $5.5 million related to PARADIGM Trial preparatory activities, including clinical costs associated with the enrollment of additional patients and the scaling of our field-based clinical team, and an increase of $1.0 million related to an expansion of our medical affairs activities. These variances were partly offset by lower DurAVR® THV product research costs of $9.5 million in 2025 as we shift our focus to clinical, regulatory and manufacturing activities ahead of the PARADIGM Trial.
Selling, General and Administrative Expense
Selling, general and administrative expenses in 2025 were $26.1 million, a decrease of $2.1 million (7%) compared to $28.2 million in 2024, primarily due to a $0.5 million decline in stock-based payment expenses associated with directors and executive management, a $0.6 million reduction in travel and entertainment costs and $1.5 million relating to a settled claim in 2024. These variances were partly offset by a $0.5 million increase in legal, tax and other operational costs, which primarily included fees related to compliance with dual listing requirements, capital raising activities and other operational matters in 2025, and, in 2024, included costs related to re-domiciliation, the listing of our Common Stock on Nasdaq, and the completion of our initial public offering.
Other non-operating income, net
Other non-operating income, net in 2025 was $0.5 million, a decrease of $2.0 million (81%) compared to $2.4 million in 2024, primarily due to the recognition of holdback income in 2024 of $0.9 million related to a transaction with LeMaitre in 2019 for which there was no corresponding income in 2025. In 2024, $0.8 million of government grants relating to the Australian R&D Tax Incentive were recognized with no corresponding income in 2025.
Net Foreign Exchange Gains/(Losses)
Net foreign exchange losses in 2025 were $0.7 million compared to $1.4 million of net foreign exchange gains in 2024, which was primarily due to the change in foreign exchange rates on intercompany and cash balances. In 2025, the United States dollar depreciated by 8% relative to the Australian dollar. In 2024, the United States dollar appreciated by 9% relative to the Australian dollar.
Debt Issuance Costs
Debt issuance costs in 2024 were $0.5 million, primarily due to the entry into a secured convertible note facility in 2024. The convertible notes were recognized at fair value through profit or loss which resulted in the costs being expensed when incurred. We did not have a corresponding charge in 2025.
Loss on Debt Extinguishment
Loss on debt extinguishment in 2024 was $0.9 million primarily due to settlement of the secured convertible note facility at a loss. We did not have a corresponding charge in 2025.
Net Income/(Loss) Attributable to Non-Controlling Interests
Net loss attributable to non-controlling interests (“NCI”) was $0.1 million in 2025, a decrease of $0.4 million (125%) compared to a $0.3 million income in 2024. The movement reflects the application of the hypothetical liquidation at book approach to measure the NCI interest.
Liquidity and Capital Resources
Capital Requirements and Sources of Liquidity
We have experienced recurring operating losses and negative cash flows from operating activities since inception. As of December 31, 2025 and December 31, 2024, we had an accumulated deficit of $370.5 million and $276.4 million, respectively.
In recent years, our operations have primarily been financed through the issuance of capital stock, including in our initial public offering, the 2025 Private Placement, as well as through convertible notes, sales of regenerative tissue products and R&D tax incentives from the Australian government. We have also generated additional funding through interest earned on cash deposits. As of December 31, 2025 and December 31, 2024, we had cash and cash equivalents of $12.6 million and $70.5 million, respectively. As of December 31, 2025 and December 31, 2024, we had capital commitments of $2.2 million and $1.4 million, respectively, relating to the lease of properties, and we did not have any other material capital expenditure commitments or contingent liabilities as of December 31, 2025.
Subsequent to year-end, we strengthened our capital position through a public equity offering and a concurrent private placement. Specifically, we completed a public offering of 40,000,000 shares of Common Stock for gross proceeds of $230 million before underwriting discounts, commissions and other transaction costs, including the underwriters' option to purchase additional shares, and a private placement to Medtronic plc (through a wholly owned subsidiary) of 15,652,173 shares of Common Stock for gross proceeds of $90 million before transaction costs. Based on the resulting increase in available liquidity, we expect our current cash on hand to be sufficient to fund our operations for at least 12 months following December 31, 2025. However, our assessment of the period of time through which our financial resources will be adequate to support our operations involves risks and uncertainties, and actual results could vary materially from our forecasts.
We anticipate that we will require substantial additional funds in order to achieve our long-term goals and complete the R&D of our current products. We do not expect to generate significant revenue until we obtain regulatory approval to market and sell our products and sales of our products have commenced. We therefore expect to continue to incur substantial losses in the near future.
Our future capital requirements are difficult to forecast and will depend on many factors, including:
•
the scope, results and timing of clinical trials;
•
the costs of preparing and completing the PARADIGM Trial of our DurAVR® THV System;
•
the costs and time required to obtain premarket approval from the FDA for our DurAVR® THV System; and
•
the costs of establishing marketing, sales and distribution capabilities.
We may seek to raise any necessary capital through a combination of public or private equity offerings or debt financings. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we decide to raise capital by issuing equity securities, the issuance of such equity securities may result in dilution to our existing stockholders. See “Risk Factors - Future equity financings and sales by existing holders could adversely affect the voting power or value of our Common Stock.” We cannot give any assurance that we will be successful in completing any financings or that any such equity or debt financing will be available to us if and when required or on satisfactory terms.
Cash Flows
The following table summarizes our primary sources and uses of cash for the periods presented (in thousands, except percentages):
| Year Ended December 31, | ||||||||||||
| 2025 | 2024 | % Change | ||||||||||
|
Net Cash provided by (used in):
|
||||||||||||
|
Operating activities
|
$ | (77,803 | ) | $ | (61,241 | ) | 27 | % | ||||
|
Investing activities
|
(596 | ) | (2,280 | ) | (74 | )% | ||||||
|
Financing activities
|
20,555 | 112,833 | (82 | )% | ||||||||
|
Effect of exchange rate movements on cash, cash equivalents and restricted cash
|
(38 | ) | 57 | (167 | )% | |||||||
|
Net change in cash, cash equivalents and restricted cash
|
$ | (57,882 | ) | $ | 49,369 | (217 | )% | |||||
Operating Activities
Net cash used in operating activities during 2025 was $77.8 million, an increase of $16.6 million (27%), compared to $61.2 million in 2024, primarily due to an increase in R&D expenses relating to the upscaling of manufacturing capabilities including process design and validation activities, preparatory activities linked to the PARADIGM Trial, including clinical costs associated with the enrollment of additional patients and an increase in employee compensation primarily linked to an increase in headcount. This increase was partly offset by a reduction in selling, general and administrative expenses relating to lower marketing spending, a decline in travel and entertainment costs and a decrease in legal, tax and compliance costs linked to a reduction in costs from 2024, which included our re-domiciliation, the listing of our Common Stock on Nasdaq and the completion of our initial public offering, relative to 2025, which included additional costs related to compliance with dual listing requirements and other operational matters. Additionally, $0.6 million of proceeds relating to the Australian R&D Tax Incentive were received in 2025, a decrease of $0.4 million compared with 2024.
Investing Activities
Net cash used in investing activities in 2025 was $0.6 million, a decrease of $1.7 million (74%), compared to $2.3 million in 2024. This decrease primarily reflects the receipt of $1.4 million deferred proceeds from LeMaitre in 2025 relating to the 2019 sale of distribution rights, for which there was no corresponding inflow in 2024. Additionally, investing cash outflows for plant and equipment decreased by $0.3 million compared with 2024.
Financing Activities
Net cash provided by financing activities in 2025 was $20.6 million, a decrease of $92.3 million (82%), compared to $112.8 million in 2024. Net proceeds from the issuance of Common Stock, net of transaction costs, were $23.0 million in 2025, down from $115.7 million in 2024, which included our initial public offering. In 2025, we also paid $1.2 million in tax withholding obligations associated with equity award settlements, whereas no such payments occurred in 2024. No convertible notes were issued or redeemed in 2025, compared with $5.0 million of proceeds and $7.2 million of repayments of debt instruments in 2024. Additionally, $1.2 million of cash outflows were associated with supplier financing arrangements to fund our annual insurance premiums in 2025, an increase of $0.5 million compared with 2024.
Contractual Obligations and Commitments
Leases
We lease laboratory facilities and offices. The leases typically include options to renew at which time the lease payments are subject to market adjustments and/or set price increases. Extension and termination options are included in a number of the leases to allow for flexibility in terms of corporate growth and managing the assets used in our operations. The leases expire between 2026 and 2030 and some include options to extend. At December 31, 2025, we had contractual commitments (on an undiscounted basis) for property leases of $2.8 million, which were recognized at $2.2 million.
Commitments
At December 31, 2025, we had commitments to purchase $0.1 million of plant and equipment.
Off-Balance Sheet Arrangements
We currently do not have, and did not have during the periods presented, any off-balance sheet arrangements.
Critical Accounting Policies and Estimates
We have used various accounting policies to prepare the consolidated financial statements in accordance with generally accepted accounting principles in the United States (“U.S. GAAP”). Our significant accounting policies and estimates are more fully described in note 2 to our audited consolidated financial statements.
The preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make judgments, estimates and assumptions that affect the reported amounts in the consolidated financial statements and accompanying notes thereto. Management continually evaluates its judgments and estimates in relation to assets, liabilities, contingent liabilities, revenue and expenses. Management bases its judgments, estimates and assumptions on historical experience and on other various factors, including expectations regarding future events that management believes to be reasonable under the circumstances. Actual results could differ from those estimates due to risks and uncertainties and may be material.
Management has discussed the development and selection of these critical accounting estimates with the Audit and Risk Committee and our Board. In addition, there are other items within our financial statements that require estimation but are not deemed critical. Changes in estimates used in these and other items could have a material impact on our financial statements.
We believe that the following discussion addresses our most critical accounting policies and estimates, which are those that are most important to the portrayal of our financial condition and results of operations and require management’s most difficult, subjective and complex judgments.
Stock-Based Payments
Equity-settled stock-based compensation benefits are provided to employees, directors and consultants in exchange for the rendering of services. We measure and recognize compensation expense for all stock-based awards based on estimated fair values determined at grant date. Fair value is determined using the Black-Scholes model which requires various inputs including the exercise price and share price at grant date, plus other highly judgmental assumptions, such as share price volatility, risk-free interest rate, and the expected option term. For options with service conditions, the expense is recognized over the service period. Stock-based compensation expense is recorded net of estimate forfeitures which is based on historical employee attrition rates. Forfeitures are estimated at the time of grant and we reassess the probability of vesting at each quarter end and adjust the stock-based compensation expense based on its probability assessment. Judgment is required in estimating which stock options will ultimately be forfeited. If actual results differ significantly from these estimates, stock-based compensation expense and our results of operations would be impacted.
The following key assumptions were used in valuing stock-based payments:
•
Risk-free interest rates for awards granted during the year ended December 31, 2025 were based on U.S. government bond yields aligned with the expected life of the securities, ranging from 3.47% to 4.41%. The rates for the year ended December 31, 2024 were based on Australian government bond yields, ranging from 3.56% to 4.48%.
•
Expected share price volatility for the year ended December 31, 2025 ranged from 60.0% to 77.5%, and for the year ended December 31, 2024 ranged from 40.0% to 65.5%. The volatility assumptions were based on our historic share price volatility over a period consistent with the expected life of the awards and adjusted for any anticipated future changes using publicly available information.
Consolidation of VIEs
We consolidate a VIE when the reporting entity (a) has an economic interest in another legal entity (known as a “variable interest”) that conveys more than insignificant exposure to potential losses of or benefits from the other legal entity; and (b) has power over the most significant economic activities of the legal entity. There is significant judgment over the analysis to determine whether an entity is a VIE, to determine whether we have a variable interest and to determine whether we are the primary beneficiary of a VIE.
We determined that v2vmedtech is a VIE and that we are the primary beneficiary of v2vmedtech. This determination is based on our having both power over the most significant activities of v2vmedtech, primarily through holding a majority of the positions on v2vmedtech’s board of directors (although v2vmedtech’s non-ATPL shareholder representative on the v2vmedtech board of directors presently maintains certain veto rights), controlling the appointment of the chief executive officer and chief financial officer roles, being the exclusive partner to develop v2vmedtech’s products, and benefits through equity ownership.
Emerging Growth Company and Smaller Reporting Company Status
We are an “emerging growth company,” (“EGC”), as defined in the JOBS Act. We will remain an EGC until the earliest of: (i) the last day of the fiscal year following the fifth anniversary of the consummation of our initial public offering; (ii) the last day of the fiscal year in which we have total annual gross revenue of at least $1.235 billion; (iii) the last day of the fiscal year in which we are deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our Common Stock (including Common Stock represented by CDIs) held by non-affiliates exceeded $700.0 million as of the last business day of the second fiscal quarter of such year; or (iv) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period. An EGC may take advantage of specified reduced reporting requirements and is relieved of certain other significant requirements that are otherwise generally applicable to public companies. As an EGC:
•
we will avail ourselves of the exemption from the requirement to obtain an attestation and report from our independent registered public accounting firm on the assessment of our ICFR pursuant to the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”);
•
we will provide less extensive disclosure about our executive compensation arrangements; and
•
we will not require non-binding, advisory stockholder votes on executive compensation or golden parachute arrangements.
In addition, the JOBS Act provides that an EGC can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an EGC to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this extended transition period for any new or revised accounting standards during the period in which we remain an EGC.
As a result, the information that we provide to our investors may be different than what you might receive from other public reporting companies. However, we may adopt certain new or revised accounting standards early.
We are also a “smaller reporting company” (“SRC”), as defined in the Exchange Act. We may continue to be a SRC even after we are no longer an EGC. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as the market value of our Common Stock (including Common Stock represented by CDIs) held by non-affiliates is less than $250.0 million measured on the last business day of our second fiscal quarter, or our annual revenue is less than $100.0 million during the most recently completed fiscal year and the market value of our Common Stock (including Common Stock represented by CDIs) held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.
As a SRC we will present only two years of audited annual financial statements, plus any required unaudited interim condensed financial statements, and related management’s discussion and analysis of financial condition and results of operations.
New Accounting Standards Not Yet Adopted
New accounting pronouncements are issued by the Financial Accounting Standards Board (“FASB”) and adopted by us as of the specified effective date. If not explicitly addressed otherwise, we believe that the recently issued standards, which have not yet taken effect, will not materially affect our present or near future financial statements.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740) Improvements to Income Tax Disclosures. ASU 2023-09 intends to enhance income tax disclosures to address investor requests for more information about the tax risks and opportunities present in an entity’s worldwide operations. The ASU’s two primary enhancements will require further disaggregation for existing disclosures for the effective tax rate reconciliation and income taxes paid. This ASU is effective January 1, 2026 for entities applying the EGC extended transition period (i.e., non‑Public Benefit Entity (“PBE’) effective dates). We have evaluated the effect of adopting this accounting guidance and will include the new required disclosures in future filings as needed.
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses. This ASU is effective January 1, 2027. ASU 2024-03 will require us to disclose the amounts of purchases of inventory, employee compensation, depreciation and intangible asset amortization, as applicable, included in certain expense captions in the Consolidated Statements of Operations, as well as qualitatively describe remaining amounts included in those captions. ASU 2024-03 will also require us to disclose both the amount and our definition of selling expenses. Adoption of this ASU will not impact the recognition or measurement of amounts in our consolidated financial statements and will result only in additional disclosure requirements.
In December 2025, the FASB issued ASU 2025‑10, Government Grants (Topic 832): Accounting for Government Grants Received by Business Entities. This ASU is effective January 1, 2030 for entities applying the EGC extended transition period (i.e., non‑PBE effective dates). ASU 2025‑10 establishes authoritative guidance on the recognition, measurement, presentation, and disclosure of government grants received by business entities. The ASU incorporates principles from IAS 20, Accounting for Government Grants and Disclosure of Government Assistance, with certain targeted refinements, and expands ASC 832 beyond disclosure‑only requirements to include a comprehensive accounting model. We have historically accounted for government grants by applying the principles of IAS 20 by analogy, which the ASU substantially aligns with. As a result, consistent with the SEC’s expectations for entities already applying IAS 20 principles, we do not expect the adoption of ASU 2025‑10 to have a significant impact on our accounting policies or consolidated financial statements.
We are a smaller reporting company, as defined
by Rule 12b-2 under the Securities and Exchange Act of 1934, as amended and in
Item 10(f)(1) of Regulation S-K, and are not required to provide the
information under this item.
ANTERIS TECHNOLOGIES GLOBAL CORP.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2025
| Page | |
| 68 | |
| 69 | |
| 70 | |
| 71 | |
| 72 | |
| 73 | |
| 74 |
To the Stockholders and Board of Directors
Anteris Technologies Global Corp.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Anteris Technologies Global Corp. and subsidiaries (the Company) as of December 31, 2025 and 2024, the related consolidated statements of operations, comprehensive loss, stockholders’ equity, and cash flows for each of the years in the two‑year period ended December 31, 2025, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2025 and 2024, and the results of its operations and its cash flows for each of the years in the two‑year period ended December 31, 2025, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ |
| |
| We have served as the Company’s auditor since 2024. |
| |
| February 26, 2026 |
ANTERIS TECHNOLOGIES GLOBAL CORP.
(In thousands of U.S. dollars, except per share information)
| Years ended December 31, | ||||||||||||
| Note | 2025 $ | 2024 $ | ||||||||||
|
Net sales
|
||||||||||||
|
Costs and expenses:
|
||||||||||||
|
Cost of products sold
|
( | ) | ( | ) | ||||||||
|
Research and development expense
|
( | ) | ( | ) | ||||||||
|
Selling, general and administrative expense
|
( | ) | ( | ) | ||||||||
|
Operating loss
|
( | ) | ( | ) | ||||||||
|
Other non-operating income, net
|
5 | |||||||||||
|
Interest and amortization of debt discount and expense
|
( | ) | ( | ) | ||||||||
|
Net foreign exchange (losses)/gains
|
( | ) | ||||||||||
|
Debt issuance costs
|
11 | ( | ) | |||||||||
|
Loss on debt extinguishment
|
11 | ( | ) | |||||||||
|
Fair value movement of derivatives
|
( | ) | ||||||||||
|
Loss before income taxes from continuing operations
|
( | ) | ( | ) | ||||||||
|
Income tax (expense)/benefit
|
6 | |||||||||||
|
Loss after income tax
|
( | ) | ( | ) | ||||||||
|
Total (loss)/gain is attributable to:
|
||||||||||||
|
Non-controlling interests
|
( | ) | ||||||||||
|
Stockholders of the Company
|
( | ) | ( | ) | ||||||||
| ( | ) | ( | ) | |||||||||
|
Share information
|
||||||||||||
|
Basic and diluted loss per share ($ per share)
|
14 | ( | ) | ( | ) | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
(In thousands of U.S. dollars)
| Years ended December 31, | ||||||||
|
2025
$ |
2024
$ | |||||||
|
Loss after income tax
|
( | ) | ( | ) | ||||
|
Other comprehensive income/(loss), net of tax:
|
||||||||
|
Foreign currency translation adjustments
|
( | ) | ||||||
|
Other comprehensive income/(loss) for the year, net of tax
|
( | ) | ||||||
|
Total comprehensive loss
|
( | ) | ( | ) | ||||
|
Total comprehensive loss is attributable to:
|
||||||||
|
Non-controlling interests
|
( | ) | ||||||
|
Stockholders of the Company
|
( | ) | ( | ) | ||||
| ( | ) | ( | ) | |||||
The accompanying notes are an integral part of these consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
(In thousands of U.S. dollars, except share quantities)
| December 31, | ||||||||||||
| Note |
2025 $ |
2024 $ | ||||||||||
|
ASSETS
|
||||||||||||
|
Current Assets
|
||||||||||||
|
Cash, cash equivalents and restricted cash
|
||||||||||||
|
Accounts receivable from customers, net of allowances
|
||||||||||||
|
Inventories
|
||||||||||||
|
Prepaid expenses
|
||||||||||||
|
Other current assets
|
9 | |||||||||||
|
Total Current Assets
|
||||||||||||
|
Non-Current Assets
|
||||||||||||
|
Plant and equipment, net
|
7 | |||||||||||
|
Operating lease right-of-use assets, net
|
8 | |||||||||||
|
Intangible assets, net
|
||||||||||||
|
Total Non-Current Assets
|
||||||||||||
|
TOTAL ASSETS
|
||||||||||||
|
LIABILITIES
|
||||||||||||
|
Current Liabilities
|
||||||||||||
|
Accounts payable
|
||||||||||||
|
Accrued and other liabilities
|
10 | |||||||||||
|
Current portion of operating lease liabilities
|
8 | |||||||||||
|
Current portion of debt obligations
|
||||||||||||
|
Total Current Liabilities
|
||||||||||||
|
Non-Current Liabilities
|
||||||||||||
|
Operating lease liabilities
|
8 | |||||||||||
|
Long-term debt obligations
|
- | |||||||||||
|
Other liabilities
|
10 | |||||||||||
|
Total Non-Current Liabilities
|
||||||||||||
|
TOTAL LIABILITIES
|
||||||||||||
|
COMMITMENTS AND CONTINGENCIES
|
19 | |||||||||||
|
STOCKHOLDERS’ EQUITY
|
||||||||||||
| Common Stock, $ |
13 | |||||||||||
| Preferred stock, $ |
||||||||||||
|
Additional paid in capital
|
||||||||||||
|
Accumulated other comprehensive loss
|
17 | ( | ) | ( | ) | |||||||
|
Accumulated deficit
|
( | ) | ( | ) | ||||||||
|
TOTAL STOCKHOLDERS’ EQUITY
|
( | ) | ||||||||||
|
Non-controlling interests
|
16 | ( | ) | ( | ) | |||||||
|
TOTAL EQUITY (DEFICIT)
|
( | ) | ||||||||||
|
TOTAL LIABILITIES AND EQUITY
|
||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
(In thousands of U.S. dollars, except share quantities)
|
| Common Stock |
Additional
Paid
in Capital
$
|
Accumulated
Other
Comprehensive
Loss
$
|
Accumulated
Deficit
$
|
Total
Stockholders’
Equity
$
| |||||||||||||||||||||||||||
|
Shares
Quantity
|
Par Value
$
|
Non-
controlling interests
$
|
Total Equity
(Deficit)
$
| |||||||||||||||||||||||||||||
|
Balance at December 31, 2023
|
( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
|
(Loss)/Gain after income tax
|
- | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||||
|
Other comprehensive loss
|
- | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||||
|
Common Stock issued
|
||||||||||||||||||||||||||||||||
|
Stock-based compensation
|
- | |||||||||||||||||||||||||||||||
|
Balance at December 31, 2024
|
( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
|
(Loss)/Gain after income tax
|
- | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||||
|
Other comprehensive income
|
- | |||||||||||||||||||||||||||||||
|
Common Stock issued
|
||||||||||||||||||||||||||||||||
|
Stock-based compensation
|
- | |||||||||||||||||||||||||||||||
| Balance at December 31, 2025 | ( | ) | ( | ) | ( | ) | ( | ( | ) | |||||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
(In thousands of U.S. dollars)
| Years ended December 31, | ||||||||||||
| Note |
2025
$ |
2024 $ | ||||||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES
|
||||||||||||
|
Loss after income tax
|
( | ) | ( | ) | ||||||||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Depreciation and amortization expense
|
||||||||||||
|
Equity-settled stock-based compensation
|
||||||||||||
|
Loss on debt extinguishment
|
||||||||||||
|
Debt issuance costs
|
||||||||||||
|
Net foreign exchange losses/(gains)
|
( | ) | ||||||||||
|
Other items
|
( | ) | ||||||||||
|
Change in operating assets and liabilities:
|
||||||||||||
|
Accounts receivable, prepayments and other assets
|
( | ) | ( | ) | ||||||||
|
Inventories
|
( | ) | ||||||||||
|
Accounts payable, accrued and other liabilities
|
||||||||||||
|
NET CASH USED IN OPERATING ACTIVITIES
|
( | ) | ( | ) | ||||||||
|
CASH FLOWS FROM INVESTING ACTIVITIES
|
||||||||||||
|
Acquisition of plant and equipment
|
( | ) | ( | ) | ||||||||
|
Acquisition of intangible assets
|
( | ) | ||||||||||
|
Deferred proceeds from sale of distribution rights
|
5 | |||||||||||
|
NET CASH USED IN INVESTING ACTIVITIES
|
( | ) | ( | ) | ||||||||
|
CASH FLOWS FROM FINANCING ACTIVITIES
|
||||||||||||
|
Net proceeds from share issues
|
13 | |||||||||||
|
Share issue transaction costs
|
13 | ( | ) | ( | ) | |||||||
|
Tax withholding paid on stock option exercises
|
13 | ( | ) | |||||||||
|
Proceeds from issuance of convertible notes
|
11 | |||||||||||
|
Repayment of debt
|
11 | ( | ) | ( | ) | |||||||
|
Debt issuance costs paid
|
( | ) | ||||||||||
|
Cash settlement of contingent options on debt
|
( | ) | ||||||||||
|
Principal payments on finance lease obligations
|
( | ) | ( | ) | ||||||||
|
NET CASH PROVIDED BY FINANCING ACTIVITIES
|
||||||||||||
|
Effect of exchange rate movements on cash, cash equivalents and restricted cash
|
( | ) | ||||||||||
|
CASH, CASH EQUIVALENTS AND RESTRICTED CASH
|
||||||||||||
|
Net change during the year
|
( | ) | ||||||||||
|
Balance at beginning of year
|
||||||||||||
|
Balance at end of year
|
||||||||||||
|
SUPPLEMENTAL CASH FLOW INFORMATION
|
||||||||||||
|
Cash received for research and development tax incentive
|
||||||||||||
| Operating cash outflows relating to operating leases | ( | ) | ( | ) | ||||||||
|
Non-cash additions to right-of-use assets and lease liabilities
|
||||||||||||
|
Non-cash issue of shares/warrants to consultants for services provided
|
||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
FOR THE YEAR ENDED DECEMBER 31, 2025
1.
DESCRIPTION OF BUSINESS
Anteris Technologies Global Corp. (“ATGC,” “Anteris,” “Company,” “we,” “us,” or “our”) was incorporated in Delaware on January 29, 2024. ATGC was formed for the purpose of reorganizing the operations of Anteris Technologies Pty Ltd (“ATPL”, formerly Anteris Technologies Ltd), an Australian public company originally registered in Western Australia, Australia and listed on the Australian Securities Exchange (“ASX”), into a structure whereby the ultimate parent company would be a Delaware corporation (the “reverse recapitalization”).
On December 16, 2024, the Company received all the issued and outstanding shares of ATPL pursuant to a scheme of arrangement under Australian law between ATPL and its shareholders (the “Scheme”) under Part 5.1 of the Australian Corporations Act 2001 (Cth) (the “Corporations Act”). Contemporaneously with implementation of the Scheme, ATPL cancelled all existing options it had outstanding in exchange for ATGC issuing replacement options to acquire shares of ATGC’s common stock, par value $0.0001 per share (“Common Stock”) pursuant to a scheme of arrangement between ATPL and its option holders (the “Option Scheme”) under Part 5.1 of the Corporations Act.
Prior to completion of the reverse recapitalization, ATGC had no business or operations and following completion of the reverse recapitalization, the business and operations of ATGC consist solely of the business and operations of ATPL and its subsidiaries. As a result of the reverse recapitalization, ATGC became the parent company of ATPL, and for financial reporting purposes the historical financial statements of ATPL became the historical financial statements of ATGC as a continuation of the predecessor.
On December 16, 2024, the Company completed the reverse recapitalization. On December 16, 2024, following the reverse recapitalization, the Company completed an initial public offering (“IPO”) of 14,800,000 shares of Common Stock.
ATGC’s principal activities consist of:
●
Continued research and development (“R&D”) of development of the DurAVR® THV, consisting of a single-piece biomimetic valve made with our primary ADAPT® tissue-enhancing technology and deployed with our ComASUR® balloon-expandable Delivery System, to address unmet medical needs in the treatment of aortic stenosis. The DurAVR® THV, with its single piece, native-shaped biomimetic design is built to mimic the performance of a healthy aortic valve and to restore normal laminar blood flow. This new class of technology can be used to treat new aortic stenosis patients and to treat aortic stenosis patients where their current bioprosthetic aortic valve is failing (“valve-in-valve”).
●
Generating and compiling clinical data through the randomized global pivotal study (the “PARADIGM Trial”), following receipt of Investigational Device Exemption (“IDE”) approval from the U.S. Food and Drug Administration (“FDA”) in November 2025, including staged authorization to enroll the first 200 patients. Data from the PARADIGM Trial is intended to support a premarket approval application (“PMA”) in the United States and a parallel CE Mark approval in Europe, key milestones on the path to commercialization.
●
The co-development with v2vmedtech, inc. (“v2vmedtech”), of an innovative heart valve repair device for the minimally invasive treatment of mitral and tricuspid valve regurgitation (also known as a leaky valve).
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
(a)
Basis of presentation
The consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”). These policies have been consistently applied to all the years presented, unless otherwise stated.
Unless noted otherwise, all dollar amounts are in thousands of United States dollars (“U.S. dollars” or “$”). Some amounts may not reconcile due to rounding.
For the year ended December 31, 2024, the consolidated financial statements reflect the consolidated results of operations, comprehensive loss, cash flows, and changes in equity of ATPL and its wholly-owned subsidiaries for the period of January 1, 2024 up to December 16, 2024, the closing date of the reverse recapitalization (the “Closing Date”), and the consolidated results of operations, comprehensive income/(loss), cash flows, and changes in stockholders’ equity of ATGC and its consolidated subsidiaries, including ATPL, for the period of December 16, 2024 through December 31, 2024. The consolidated balance sheet at December 31, 2024 presents the financial condition of the Company and its consolidated subsidiaries, including ATPL.
In accordance with ASC 805, Business Combinations, ATPL’s historical equity has been retrospectively restated for all periods up to the Closing Date to reflect the number of shares of Common Stock issued to legacy ATPL shareholders in connection with the reverse recapitalization. Additionally, the par value of Common Stock has been restated to align with the post-transaction capital structure.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
The Company is an emerging growth company (“EGC”), as defined in Section 2(a) of the Securities Act of 1933 (the “Securities Act”), as modified by the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), which permits the Company to utilize an extended transition period to comply with new or revised accounting standards applicable to public companies.
(b)
Principles of consolidation
The consolidated financial statements include the accounts of ATGC, its wholly-owned subsidiaries, entities for which the Company has a controlling financial interest, as well as any variable interest entities (“VIEs”) for which ATGC has been determined to be the primary beneficiary. ATGC and its subsidiaries together are referred to in these financial statements as the “Group”.
Subsidiaries are all those entities over which the Group has control. Control is the power to govern the financial and operating policies of an entity. All subsidiaries of ATGC have a reporting year end of December 31.
Intercompany transactions, balances and unrealized gains or losses on transactions between entities in the Group are eliminated.
(c)
Use of estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make judgments, estimates and assumptions that affect the reported amounts in the consolidated financial statements and accompanying notes. Management continually evaluates its judgments and estimates in relation to assets, liabilities, contingent liabilities, revenue and expenses. Management bases its judgments, estimates and assumptions on historical experience and on other various factors, including expectations of future events that management believes to be reasonable under the circumstances. Actual results could differ from those estimates due to risks and uncertainties and may be material.
Management has discussed the development and selection of these critical accounting estimates with the Company’s Audit and Risk Committee and Board of Directors (the “Board”). In addition, there are other items within the financial statements that require estimation but are not deemed critical as defined above. Changes in estimates used in these and other items could have a material impact on the financial statements.
Significant items subject to such estimates and assumptions include, but are not limited to the following:
●
Stock-based compensation fair value inputs: Fair value is estimated using option pricing models, including the Black-Scholes option pricing model (the “Black-Scholes model”). The model requires various inputs including the exercise price and share price at grant date, plus other judgmental assumptions, such as share price volatility, risk-free interest rate, and the expected option term.
(d)
Foreign currency translation
The financial statements are presented in United States dollars, which is ATGC’s reporting currency.
Foreign currency transactions
Foreign currency transactions are translated using the average monthly currency exchange rates in effect during the period. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at period-end exchange rates of monetary assets and liabilities denominated in foreign currencies are included as operating income or expense in the consolidated statements of operations.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Conversion to presentation currency
The assets and liabilities of non-U.S. dollar functional currency entities are translated into U.S. dollars using period-end exchange rates, and the revenues and expenses of those entities are translated into U.S. dollars using the average exchange rates, which approximates the rate at the date of the transaction. Equity accounts are translated at historical rates, except for the change in retained earnings during the year, which is the result of the income statement translation process. The cumulative translation adjustments associated with the net assets of foreign subsidiaries are recorded in accumulated other comprehensive loss in the consolidated statements of comprehensive loss and stockholders’ equity.
The determination of the functional and reporting currency of each Group company is based on the primary currency in which the Group company operates. ATGC and Anteris Technologies Corporation have the U.S. dollar (“USD” or “$”) as their functional currency, ATPL and the Australian subsidiaries’ use the Australian dollar (“AUD” or “A$”) as their functional currency, and another subsidiary has the Swiss franc (“CHF”) as its functional currency.
(e)
Net sales
Net sales from the sale of goods, which is primarily ADAPT® tissue, are recognized at a point in time when the performance obligation is satisfied, typically being upon delivery to the customer’s premises when control of the goods transfers to the customer. Revenue is recognized at an amount which reflects the consideration to which the Group expects to be entitled in exchange for those goods.
The Company generates its revenue from direct product sales and typically does not have any significant unusual payment terms beyond 30 days in its contracts with customers.
Revenue recognition is determined by considering sales rebates and returns, which are assessed through sales terms, historical data, and trend analysis. When estimating rebates, the Company takes into account factors such as the stated rebate rates, trending volumes and other relevant information. Adjustments to rebates and returns reserves are recorded by the Company as either revenue increases or decreases. The Company offers warranties on its tissue and valves that they conform to the specifications, fit for their intended purpose, and do not have material defects.
Taxes assessed by a governmental authority that are both imposed on specific revenue producing transactions and collected by the Company from customers (for example, sales, use, value added, and some excise taxes) are not included in revenue.
(f)
Other income
Interest income
Interest income is recognized as interest accrues using the effective interest method. This is a method of calculating the amortized cost of a financial asset and allocating the interest income over the relevant period using the effective interest rate, which is the rate that exactly discounts contractual future cash receipts through the expected life of the financial asset to the net carrying amount of the financial asset.
Other income
Other income is recognized when it is received or when the right to receive payment is established.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Research and development tax incentive income
Government grants are received under the Australian government’s research and development tax incentive program, such that a percentage of eligible R&D expenses are reimbursed by the Australian government with the incentive being recognized as other income. Government grants relating to costs incurred are recognized in the consolidated statements of operations over the periods in which the entity recognizes as expenses the related costs for which they are intended to compensate.
The research and development tax incentive income is recognized as income once the Group is satisfied that the Group has complied with the conditions attached to the tax incentives and that the tax incentives will be received. The value is estimated based on an assessment of actual and budgeted eligible R&D expenditure data for the period. Significant judgment is required in determining the amount and timing of recognition, as the grant requirements are complex.
(g)
Cash, cash equivalents and restricted cash
Cash and cash equivalents includes cash on hand, deposits held at call with financial institutions, other short-term, highly liquid investments with original maturities of three months or less that are readily convertible to known amounts of cash and which are subject to an insignificant risk of change in value. The Company’s restricted cash primarily represent funds placed in escrow related to operating leases and as collateral for a credit line. As of December 31, 2025, the restricted cash balance was $0.5 million.
(h)
Accounts receivable and other financing receivables
Accounts receivable are amounts due from customers for direct product sales in the ordinary course of business. They are generally due for settlement within 30 days and therefore all classified as current. Accounts receivable are recognized initially at the amount of consideration that is unconditional. The Company holds the accounts receivable with the objective to collect the contractual cash flows and therefore measures them subsequently at amortized cost, less impairment allowances.
An allowance is maintained for estimated losses in the collection of accounts receivable based on customer-specific analysis. The allowance is assessed by considering factors including the recent sales experience, the aging of receivables and historical collection rates. Uncollectible amounts are written-off against the allowance when it is determined that a customer account is uncollectible. Indicators that there is no reasonable expectation of recovery include, amongst others, the failure of a debtor to engage in a repayment plan with the Company, and a failure to make contractual payments for a period greater than 120 days past due. Subsequent recoveries on amounts previously written off are credited against the same line item.
Other receivables are recognized at amortized cost, less any expected loss allowance.
(i)
Inventories
Raw materials, work in progress and finished goods are stated at the lower of cost and net realizable value on a weighted average cost formula. Cost comprises direct materials and delivery costs, direct labor, import duties and other taxes, plus an appropriate proportion of variable and fixed overhead expenditure based on normal operating capacity. Costs of purchased inventory are determined after deducting rebates and discounts received or receivable.
Net realizable value is the estimated selling price in the ordinary course of business less the estimated costs of completion and the estimated costs necessary to make the sale.
The Company recognizes an inventory reserve to recognize write-downs recorded against the carrying value of inventories for items that are obsolete, damaged, nearing its expiration date, or slow-moving.
(j)
Plant and equipment
Recognition and measurement
Plant and equipment is stated at historical cost less accumulated depreciation and impairment. Historical cost includes expenditure that is directly attributable to the acquisition of the items. Additions and improvements that extend the lives of the assets are capitalized, while expenditure for repairs and maintenance are expensed as incurred.
Costs incurred in acquiring software and licenses that will contribute to future period financial benefits through revenue generation and/or cost reduction are capitalized to software and systems. Costs capitalized include external direct costs of materials and services.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
An item of plant and equipment is derecognized upon disposal or when there is no future economic benefit to the Group. Gains and losses between the carrying amount and the disposal proceeds are taken to the consolidated statements of operations. The Company assesses plant and equipment for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable.
Depreciation
Depreciation is calculated on a straight-line basis to write off the net cost of each item of plant and equipment over their expected useful lives. Leasehold improvements and plant and equipment under lease are depreciated over the unexpired period of the lease or the estimated useful life of the assets, whichever is shorter.
The residual values, useful lives and depreciation methods are reviewed, and adjusted if appropriate, at each reporting date.
Impairment of Long-lived assets
The Group assesses impairment of long-lived assets at each reporting date by evaluating conditions specific to the Group and to the asset or asset group that may lead to impairment. If an impairment trigger exists and the review indicates that the assets will not be fully recoverable based on undiscounted estimated cash flows over the remaining amortization periods, their carrying values are reduced to estimated fair market value. For the purposes of identifying and measuring impairment, long-lived assets are grouped with other assets and liabilities at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities.
(k)
Leases
The Company’s lease agreements include leases accounted for as operating leases and those accounted for as finance leases. Finance lease ROU assets are included in plant and equipment, net, and finance lease liabilities are included in current debt obligations and long-term debt on the Consolidated Balance Sheets.
The Group leases laboratory facilities and offices through operating leases. The Group leases information technology (“IT”) equipment through finance leases.
See Note 8 Leases for further information. Anteris is not a lessor in any lease arrangement.
Anteris as the Lessee
At inception of a contract, the Group assesses whether a contract is, or contains, a lease. A contract is, or contains, a lease if the contract conveys the right to control the use of an identified asset for a period of time in exchange for consideration.
At the lease commencement date, the Group recognizes a ROU asset (the right to use the leased item) and a corresponding lease liability, except for short term leases. Anteris has made an accounting policy election to apply the short-term lease election to all classes of underlying assets, being those leases which have a term of 12 months or less. The Group recognizes the lease payments associated with these leases as an expense on a straight-line basis over the lease term.
The Company determines the lease term as the noncancellable period of the lease and may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option.
Lease liabilities
For operating and finance leases, the lease liability is initially measured at the present value of the unpaid lease payments at the lease commencement date.
Lease liabilities are subsequently measured by reducing the balance to reflect the principal lease repayments made and increasing the carrying amount by the interest on the lease liability. The Group is required to remeasure the lease liability and make an adjustment to the ROU asset in the following instances:
●
the term of the lease has been modified or there has been a change in the assessment of a purchase option being exercised, in which case the lease liability is remeasured by discounting the revised lease payments using a revised discount rate; and
●
a lease contract is modified, and the lease modification is not accounted for as a separate lease, in which case the lease liability is remeasured by discounting the revised lease payments using a revised discount rate.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Lease liabilities which will be repaid within twelve months are recognized as current and the liabilities which will be repaid in excess of twelve months are recognized as non-current liabilities.
Lessees are required to discount future lease payments using the interest rate implicit in the lease or, if that rate cannot be readily determined, its incremental borrowing rate. Generally, for operating leases, the Group cannot determine the interest rate implicit in the lease because it does not have access to the lessor’s estimated residual value or the amount of the lessor’s deferred initial direct costs. Therefore, the Group generally uses its incremental borrowing rate as the discount rate for the lease. The incremental borrowing rate is the rate of interest that the Group would have to pay to borrow an amount equal to the lease payments in a similar economic environment and on a collateralized basis over a similar term.
ROU assets
The ROU asset is initially measured at cost, which comprises the initial amount of the lease liability adjusted for any lease payments made at or before the commencement date, plus any initial direct costs incurred, less any lease incentives received.
For finance leases, the ROU asset is subsequently depreciated using the straight-line method from the commencement date to the end of the lease term. Any remeasurement of the lease liability is also applied against the ROU asset value.
For operating leases, the ROU asset is subsequently measured at the amount of the remeasured lease liability, adjusted for the remaining balance of any lease incentives, accrued or prepaid rents. The carrying amount of the ROU asset approximates the present value of the remaining benefits to the Group at each measurement date.
Extension and termination options
Extension and termination options are included in a number of property operating leases across the group and are an area of judgment. In determining the lease term, management considers all facts and circumstances that create an economic incentive to exercise an extension option or not exercise a termination option. Extension options (or periods after termination options) are only included in the lease term if the lease is reasonably certain to be extended (or not terminated).
(l)
Intangibles
Intangible assets acquired as part of a business combination, other than goodwill, are initially measured at their fair value at the date of the acquisition. Intangible assets acquired separately are initially recognized at cost. Finite life intangible assets are subsequently measured at cost, less amortization and any impairment.
The method and useful lives of finite life intangible assets are reviewed at each reporting period. Changes in the expected pattern of consumption or useful life are accounted for prospectively by changing the amortization method or period.
The Group holds finite life intangible assets which are amortized on a straight-line basis with estimated useful lives.
Significant costs associated with the registration of patents and trademarks are deferred and amortized on a straight-line basis over the period of their expected benefit. The term of individual patents depends upon the legal term for patents in the countries in which they are granted. In most countries, including the United States and Australia, the patent term is 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country.
In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
Impairment of intangible assets
Intangible assets with finite lives are tested for impairment when an event occurs or circumstances change that would indicate the carrying amount of the assets or asset group may be impaired. If an impairment trigger exists, the recoverable amount of the asset is determined. Impairment indicators include, among other conditions, cash flow deficits, historic or anticipated declines in revenue or operating profit, and adverse legal or regulatory developments. If it is determined that such indicators are present and the review indicates that the assets will not be fully recoverable, based on undiscounted estimated cash flows over the remaining amortization periods, their carrying values are reduced to estimated fair market value. Estimated fair market value is determined primarily using the anticipated cash flows discounted at a rate commensurate with the risk involved.
(m)
Debt obligations
Interest-bearing debt obligations
Debt obligations are initially recognized at fair value, net of transaction costs incurred. Debt obligations are subsequently measured at amortized cost. Any difference between the proceeds (net of transaction costs) and the redemption amount is recognized in the consolidated statements of operations over the period of the borrowings using the effective interest method.
(n)
Income taxes
Income taxes are accounted for under the asset and liability method.
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and for operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those deferred tax assets and liabilities are expected to be recovered or settled. A valuation allowance is provided to reduce deferred tax assets to the amount that is more likely than not to be realized. Deferred taxes, including valuation allowances, are determined separately for each tax-paying component in each jurisdiction. The factors used to assess the likelihood of realization are future reversals of existing taxable temporary differences, carryback availability, both historical experience and the Company’s forecast of future taxable income and available tax planning strategies that could be implemented to realize the net deferred tax assets. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
Unrecognized tax benefits
The Group recognizes the effect of income tax positions only if those positions are more likely than not (greater than 50% likelihood) of being sustained upon examination by the taxing authorities, based on the technical merits of the position. Where the Group expects a tax position to be sustained, it recognizes the tax benefit as the largest amount that has a greater than 50% likelihood of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs.
The Group is subject to income taxes in the jurisdictions in which it operates. Significant judgment is required in determining unrecognized tax benefits. The Group records liabilities or makes other adjustments for unrecognized tax benefits based on the Group’s current understanding of the tax law. Where the final tax outcome of these matters is different from the carrying amounts, such differences will impact current and deferred taxes in the period in which such determination is made. The Group did not record any unrecognized tax benefits at December 31, 2025.
Inherent in determining the income tax amounts, including the valuation allowance, are judgments regarding business plans, tax planning opportunities and expectations about future outcomes. Realization of certain deferred tax assets is dependent upon generating sufficient taxable income in the appropriate jurisdiction prior to the expiration of the carryforward periods. Currently, management has recognized a valuation allowance for the amount of the deferred tax assets not supported by future reversals of existing taxable temporary differences as management believes that it is more likely than not that those deferred tax assets will not be realized.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
(o)
Stock-based payments
Equity-settled stock-based compensation benefits are provided to employees, directors and consultants in exchange for the rendering of services. Cash-settled stock-based payment transactions provide employees with the right to cash payments upon the satisfaction of vesting conditions.
The Company measures and recognizes compensation expense for all stock-based awards based on estimated fair values determined at grant date.
The fair value of the cash-settled stock-based obligation is recognized as an expense on a straight-line basis over the requisite service period, with a corresponding increase in liabilities. Upon satisfaction of the vesting conditions attached to the rights, the provision becomes a payable. The liability is remeasured at each reporting date and at settlement date based on the fair value of the rights. Any changes in the liability are recognized in profit or loss.
If equity-settled awards are modified, an additional expense is recognized, over the remaining vesting period, for any modification that increases the total fair value of the stock-based compensation benefit as of the date of modification.
Service-based stock-based compensation
Anteris offers service-based stock options and restricted stock units (“RSUs”) to employees and directors as it believes that the grant of these awards assists with attracting, motivating and aligning the employee and director interests with those of its stockholders.
Stock-based compensation expense is measured at the grant date based on the fair value of the award and is recognized as expense over the requisite service period (vesting period) on a straight-line basis. Forfeitures are estimated based on historical employee attrition rates at the time of grant and the Company reassesses the probability of vesting at each quarter end and adjusts the stock-based compensation expense based on its probability assessment. Upon exercise of stock options and upon completion of the RSU vesting conditions, the Company issues shares of Common Stock.
RSUs contingently granted to directors require stockholder approval in accordance with ASX listing rules and do not become legally binding on the Company until such approval is obtained. Under ASC 718, an award is not granted until the grantee has an enforceable right to it. When RSUs are subject to stockholder approval, this right does not exist before the vote, so no grant date is established and no expense is recognized until approval is obtained. Once stockholders approve the award, the RSUs become legally effective and the grant date is the approval date. The Company measures fair value using the share price on that date, and begins recognizing expense over the remaining service period.
Stock options and shares issued to external consultants
On occasion, the Company has granted options or shares to external consultants as consideration for services provided. Awards granted to non-employees are measured at the grant date by estimating the fair value of the equity instruments to be issued in exchange for goods or services received. The expense is recognized in the same manner as if the Company had paid cash for the services.
Fair value estimates
The fair value of stock options is determined using the Black-Scholes model which requires various inputs including the exercise price and share price at grant date, plus other highly judgmental assumptions, such as share price volatility, risk-free interest rate, and the expected option term. For stock options and RSUs with service conditions, the expense is recognized over the service period. Stock-based compensation expense is recorded net of estimate forfeitures. Judgment is required in estimating which equity securities will ultimately be forfeited. If actual results differ significantly from these estimates, stock-based compensation expense and the results of operations would be impacted.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
(p)
Earnings/Loss per share
Basic earnings (or loss) per share is computed by dividing net profit/loss by the weighted-average Common Stock outstanding during the period. Diluted earnings/loss per share is computed based on the weighted-average Common Stock outstanding plus the effect of dilutive potential Common Stock outstanding during the period calculated using the treasury stock method. Dilutive potential Common Stock includes employee equity stock options, non-vested shares, and similar equity instruments granted by the Company. Potential Common Stock equivalents have been excluded where their inclusion would be anti-dilutive.
(q)
Fair value measurement
When an asset or liability, financial or non-financial, is measured at fair value for recognition or disclosure purposes, the fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date; and assumes that the transaction will take place either in the principal market; or in the absence of a principal market, in the most advantageous market.
Valuation techniques that are appropriate in the circumstances and for which sufficient data are available to measure fair value, are used, maximizing the use of relevant observable inputs and minimizing the use of unobservable inputs.
Assets and liabilities measured at fair value are classified, into three levels, using a fair value hierarchy that reflects the significance of the inputs used in making the measurements:
●
Level 1: Quoted prices (unadjusted) in active markets for identical assets or liabilities that the entity can access at the measurement date;
●
Level 2: Inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; or
●
Level 3: Unobservable inputs for the asset or liability.
Classifications are reviewed each reporting date and transfers between levels are determined based on a reassessment of the lowest level input that is significant to the fair value measurement.
For recurring and non-recurring fair value measurements, external valuers may be used when internal expertise is either not available or when the valuation is deemed to be significant. External valuers are selected based on market knowledge and reputation. Where there is a significant change in fair value of an asset or liability from one period to another, an analysis is undertaken, which includes a verification of the major inputs applied in the latest valuation and a comparison, where applicable, with external sources of data.
(r)
Other liabilities
The Company records a liability in the consolidated financial statements where it is probable that a liability has been incurred, and the amount may be reasonably estimated. If the reasonable estimate of a probable loss is a range, and no amount within the range is a better estimate than any other, the minimum amount of the range is accrued. If a loss is reasonably possible but not probable, and may be reasonably estimated, the estimated loss or range of loss is disclosed.
Lease asset retirement obligation
The lease asset retirement obligation relates to the removing of leasehold improvements including laboratories, clean rooms and office spaces and returning the premises to their original condition in accordance with the lease agreements. The calculation of this obligation requires assumptions such as closure dates and cost estimates. The amount recognized for each site is periodically reviewed and updated based on the facts and circumstances available at the time.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
(s)
Employee benefits
Liabilities for employment benefits, which include wages and salaries, bonuses, post-employment benefits, annual leave and long-term service benefits expected to be settled within 12 months of the reporting date are measured at the amounts expected to be paid when the liabilities are settled.
The liability for annual leave and long-term service benefits not expected to be settled within 12 months of the reporting date are measured as the present value of expected future payments to be made in respect of services provided by employees up to the reporting date using the projected unit credit method. Consideration is given to expected future wage and salary levels, experience of employee departures and periods of service. Expected future payments are discounted using market yields at the reporting date on national government bonds with terms to maturity and currency that match, as closely as possible, the estimated future cash outflows.
Defined contribution savings plans
Contributions to defined contribution plans are expensed in the period in which they are incurred.
Australian employees are entitled to contributions to defined contribution plans (superannuation) at 12.5% of the participant’s annual eligible compensation (post July 1, 2025) subject to certain contribution caps. The rate has increased by 0.5% annually for the past four years. The net expense related to these plans was $0.9 million and $0.7 million in fiscal years 2025 and 2024, respectively.
The Company’s employees in the United States are eligible to participate in a qualified defined contribution plan. Employees receive 3 % employer contributions limited by the eligible compensation threshold.
(t)
Research and development expenses
Research and development expenses are recognized in the consolidated statements of operations in the period in which they are incurred. R&D costs include costs of research, engineering, and technical activities undertaken to develop new products or services or to significantly improve existing products or manufacturing processes. They also include pre-approval regulatory and clinical trial expenses; inventory costs incurred in jurisdictions where regulatory approval has not yet been obtained; and services and supplies associated with clinical studies, registries and sponsored research. These costs include direct salary and employee benefit-related costs for R&D personnel, costs for materials used and costs for outside services.
(u)
Variable Interest Entities
Under ASC 810 Consolidation (“ASC 810”), when the Company obtains an economic interest in an entity, it evaluates the entity to determine if it should be deemed a VIE, and, if so, whether the Company is the primary beneficiary and is therefore required to consolidate the VIE, based on significant judgment whether the Company (i) has the power to direct the activities that most significantly impact the economic performance of the VIE and (ii) has the obligation to absorb losses or the right to receive benefits of the VIE that could potentially be significant to the VIE.
On an ongoing basis, the Company re-evaluates the VIE assessment based on potential changes in facts and circumstances, including but not limited to, the stockholder loans to the entity and the execution of any future significant agreements between the entity and its stockholders and/or other third parties.
(v)
Segment reporting
Segment information is presented using a management approach, meaning that segment information is provided on the same basis as information is used for internal reporting purposes by the chief operating decision maker (“CODM”) which is the Vice Chairman and Chief Executive Officer, who makes key strategic decisions. The CODM is responsible for the allocation of resources and assessing the performance of the Group. Management has determined that the activities of the business as reviewed by the CODM are one segment, being the development and commercialization of the ADAPT® anti-calcification tissue. This is focused on the DurAVR® THV System.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
2.
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (continued)
(w)
Reverse recapitalization
In accordance with ASC 805, Business Combinations, when ATGC (or “the legal parent”) acquired ATPL (“the legal subsidiary”), the transaction was accounted for as a reverse recapitalization. The substance of the transaction was that the pre-transaction shareholders of ATPL (the accounting acquirer) had effectively obtained control of ATGC.
Under reverse recapitalization accounting, the consolidated financial statements are issued under the name of the legal parent (being ATGC) but, with the exception of stockholder’s equity, the financial statements will represent a continuation of ATPL’s (the legal subsidiary’s) financial information. ATPL’s assets and liabilities will be recognized at historical cost with no goodwill or other intangible assets recorded in the financial statements.
At the date of acquisition, the assets and liabilities of the Group were recognized and measured in the consolidated financial statements at their pre-combination carrying amounts and added to the assets and liabilities of ATGC.
The consolidated statement of stockholders’ equity for comparative periods has been restated to reflect the equity structure of ATGC, while maintaining the historical retained earnings of ATPL.
(x)
Recently Adopted Accounting Standard
In June 2022, the FASB issued ASU 2022-03, Fair Value Measurement (Topic 820) Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions. ASU 2022-03 clarifies guidance for fair value measurement of an equity security subject to a contractual sale restriction and establishes new disclosure requirements for such equity securities. This ASU was effective January 1, 2025 for entities applying the EGC extended transition period (i.e., non‑PBE effective dates). The adoption of ASU 2022-03 on January 1, 2025 did not materially impact the fair value measurement of our existing equity securities.
(y)
New Accounting Standards Not Yet Adopted
New accounting pronouncements are issued by the FASB and adopted by the Company as of the specified effective date. If not explicitly addressed otherwise, the Company believes that the recently issued standards, which have not yet taken effect, will not materially affect its present or near future financial statements.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740) Improvements to Income Tax Disclosures. ASU 2023-09 intends to enhance income tax disclosures to address investor requests for more information about the tax risks and opportunities present in an entity’s worldwide operations. The ASU’s two primary enhancements will require further disaggregation for existing disclosures for the effective tax rate reconciliation and income taxes paid. This ASU is effective January 1, 2026 for entities applying the EGC extended transition period (i.e., non‑PBE effective dates). The Company has evaluated the effect of adopting this accounting guidance and will include the new required disclosures in future filings.
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses. This ASU is effective January 1, 2027. ASU 2024-03 will require the Company to disclose the amounts of purchases of inventory, employee compensation, depreciation and intangible asset amortization, as applicable, included in certain expense captions in the Consolidated Statements of Operations, as well as qualitatively describe remaining amounts included in those captions. ASU 2024-03 will also require the Company to disclose both the amount and the Company’s definition of selling expenses. Adoption of this ASU will not impact the recognition or measurement of amounts in our consolidated financial statements and will result only in additional disclosure requirements.
In December 2025, the FASB issued ASU 2025‑10, Government Grants (Topic 832): Accounting for Government Grants Received by Business Entities. This ASU is effective January 1, 2030 for entities applying the EGC extended transition period (i.e., non‑PBE effective dates). ASU 2025‑10 establishes authoritative guidance on the recognition, measurement, presentation, and disclosure of government grants received by business entities. The ASU incorporates principles from IAS 20, Accounting for Government Grants and Disclosure of Government Assistance, with certain targeted refinements, and expands ASC 832 beyond disclosure‑only requirements to include a comprehensive accounting model. The Company has historically accounted for government grants by applying the principles of IAS 20 by analogy, which the ASU substantially aligns with. As a result, consistent with the SEC’s expectations for entities already applying IAS 20 principles, the Company does not expect the adoption of ASU 2025‑10 to have a significant impact on its accounting policies or consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
3.
GOING CONCERN
The consolidated financial statements have been prepared on the going concern basis, which contemplates continuity of normal business activities and realization of assets and discharges of liabilities in the ordinary course of business. As of December 31, 2025, the Group incurred a net loss of $94.2 million and had net cash outflows from operating activities of $77.8 million for the financial year ended December 31, 2025. As of that date, the Group had a cash balance of $12.6 million, a net current liability position of $5.7 million and a net liability position of $0.3 million.
Subsequent to December 31, 2025, the Company completed two equity financing transactions that significantly strengthened its liquidity position. The Company completed a public offering of 40,000,000 shares of Common Stock for gross proceeds of $230 million before underwriting discounts, commissions and other transaction costs, including the underwriters’ option to purchase additional shares. In addition, the Company completed a private placement to Medtronic plc (through a wholly owned subsidiary) of 15,652,173 shares of Common Stock for gross proceeds of $90 million before transaction costs. These transactions materially improved the Company’s available cash resources.
After considering the Group’s current cash position, the proceeds from the equity financings completed subsequent to year‑end, the Group’s forecasted cash flows, and its planned operating and investment activities, management has concluded that no substantial doubt exists regarding the Group’s ability to continue as a going concern for at least 12 months from the date these financial statements are issued.
4.
NET SALES
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Net sales from contracts with customers, at a point in time
|
||||||||
|
ADAPT® tissue products
|
||||||||
|
Total net sales
|
||||||||
5.
OTHER NON-OPERATING INCOME, NET
|
(in thousands)
|
2025 $ |
2024 $ | ||||||
|
Government grants (reversal) / income (1)
|
( | ) | ||||||
|
LeMaitre holdback income (2)
|
||||||||
|
Interest income
|
||||||||
|
Sundry income
|
||||||||
|
Total other non-operating income, net
|
||||||||
(1)
(2)
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
6.
INCOME TAXES
(a)
Income tax (expense)/benefit
The components of the (loss)/income before income taxes from continuing operations, based on tax jurisdiction, are as follows:
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
United States
|
( | ) | ( | ) | ||||
|
Australia
|
( | ) | ( | ) | ||||
|
Other international
|
||||||||
|
(Loss)/income before income taxes from continuing operations
|
( | ) | ( | ) | ||||
(b)
Deferred Tax Assets and Liabilities
Deferred taxes result from temporary differences between the amount of assets and liabilities recognized for financial reporting and income tax purposes. The significant components of the net deferred tax asset (liability) shown on the Consolidated Balance Sheets are as follows:
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Deferred tax assets
|
||||||||
|
Accrued and other liabilities
|
||||||||
|
Share issue costs
|
||||||||
|
Intangible assets
|
||||||||
|
Other capitalized costs
|
||||||||
|
Stock-based payments
|
||||||||
|
Operating lease liabilities
|
||||||||
|
Capitalized R&D
|
||||||||
|
Tax credit carryforwards
|
||||||||
|
Operating loss carryforwards
|
||||||||
|
Total deferred tax assets
|
||||||||
|
Deferred tax liabilities
|
||||||||
|
Plant and equipment
|
( | ) | ( | ) | ||||
|
Operating lease ROU assets
|
( | ) | ( | ) | ||||
|
Accounts receivable from customers, net of allowances
|
( | ) | ||||||
|
Prepaid expenses
|
( | ) | ( | ) | ||||
|
Intercompany loans
|
( | ) | ( | ) | ||||
|
Other
|
( | ) | ||||||
|
Total deferred tax liabilities
|
( | ) | ( | ) | ||||
|
Total net deferred tax assets (prior to valuation allowance)
|
||||||||
|
Valuation allowance
|
( | ) | ( | ) | ||||
|
Net deferred tax assets
|
||||||||
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
6.
INCOME TAXES (continued)
The valuation allowance of $86.8 million as of December 31, 2025 and $54.5 million as of December 31, 2024 reduces deferred tax assets to amounts that are more likely than not to be realized. This allowance primarily relates to the net operating loss carryforwards of the Group as management does not believe that it is more likely than not that these net operating losses will be utilized. The increase in the valuation allowance is primarily related to the additional net operating losses and capitalized R&D recorded during the fiscal year.
The portion of the valuation allowance for deferred tax assets for which subsequently recognized tax benefits would be applied directly to contributed capital was $2.4 million as of December 31, 2025 and $2.1 million as of December 31, 2024.
(c)
Operating loss carryforwards
Information about net operating and capital loss carryforwards at December 31, 2025 is summarized as follows:
|
(in thousands)
|
$ | |||
|
Australian net operating and capital loss carryforwards
|
||||
|
United States federal net operating loss carryforwards
|
||||
|
United States state net operating loss carryforwards
|
||||
|
Other net operating loss carryforwards
|
||||
|
Total
|
||||
Included within the Australian carryforwards disclosed above, the Australian tax consolidated group has $4.4 million of transferred losses at December 31, 2025, which are subject to loss recoupment testing and their available fraction which limits the annual rate at which the loss carryforwards may be claimed by the parent entity.
The Group’s operating loss carryforwards are subject to examination by taxing authorities specific to each jurisdiction in which they were incurred and the filing and finalization of income tax returns. The actual operating loss carryforwards available on filing of these returns may be different. The operating loss carryforwards may not be realizable in whole or in part due to the generation of significant future income being dependent on obtaining the necessary regulatory approvals, which are not in place as of December 31, 2025.
At December 31, 2025, the Company had $377.3 million of operating loss carryforwards in the United States, Australia and other international jurisdictions, of which up to $228.1 million carryforward indefinitely, with the remaining $149.2 million due to expire during fiscal years 2026 through to 2045 if not used.
An analysis of potential restrictions on our ability to utilize loss carryforwards and other income tax attributes has been performed as of December 31, 2025. No changes in ownership were identified for either the United States or Australia, therefore no restrictions on our ability to utilize our carryforwards or other income tax attributes are believed to exist as of December 31, 2025.
Following the capital raises in January 2026 (refer to Note 23 Subsequent Events for further details), further analysis of the carryforwards and other income tax attributes is being performed. If we determine we had a change in ownership it would subject the carryforwards in ATGC to a limitation under Section 382 of the Internal Revenue Code. Aggregate U.S. federal and state deferred tax assets for carryforwards attributable to ATGC are $25.6 million. No changes in ownership have been identified in Australia as a result of the equity financings completed subsequent to year-end.
On December 30, 2025, Anteris Technologies Pty Ltd, Anteris Aus Operations Pty Ltd (both Australian entities) and Anteris Corporation Ltd entered into an agreement to transfer intellectual property from Australia to the U.S., subject to the satisfaction of certain conditions precedent. If these are met, we expect some or all Australian operating loss carryforwards would be utilized to cover any capital gain arising from the transfer in the 2025 tax year.
The Group files income tax returns in a number of jurisdictions including the United States, Australia, Switzerland and Singapore. Income tax returns for all wholly-owned subsidiaries have been filed for the period ended December 31, 2024. With limited exceptions, all years prior to 2021 in Australia and 2022 in the United States are no longer subject to examination by taxing authorities.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
6.
INCOME TAXES (continued)
Operating loss carryforwards
The net operating and capital loss carryforwards, including the year they are scheduled to expire, are set out below:
|
Financial Year Ending December 31:
(in thousands)
|
$ | |||
|
2026
|
||||
|
2027
|
||||
|
2028
|
||||
|
2029
|
||||
|
2030
|
||||
|
2031
|
||||
|
2032
|
||||
|
2033
|
||||
|
2034
|
||||
|
2035
|
||||
|
2036
|
||||
|
2037 through 2045
|
||||
|
Indefinite
|
||||
|
Total
|
||||
(d)
Tax credit carryforwards
As of December 31, 2025, the Company had tax credit carryforwards of $1.327 million, comprising Australian research expenditure tax credits. These tax credit carryforwards are available to be carried forward indefinitely and do not expire.
(e)
Effective income tax rate varied from the parent’s statutory income tax rate
The Company’s reported income tax expense varied from the amount of income tax expense that would result from applying the statutory income tax rate of the parent entity, the income tax rate of the parent entity’s country of domicile. The reconciliation of the U.S. federal income tax benefit at the statutory federal income tax rate of 21
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Statutory income tax rate of the parent entity
|
% | % | ||||||
|
Domicile of parent
|
USA | USA | ||||||
|
Income tax (benefit) at the statutory income tax rate
|
( | ) | ( | ) | ||||
|
Increase / (decrease) in income tax expense resulting from:
|
||||||||
|
Non-deductible other expenses
|
||||||||
|
Non-deductible stock-based payments
|
||||||||
|
Non-assessable income
|
( | ) | ( | ) | ||||
|
Non-deductible R&D expenditure
|
||||||||
|
Foreign statutory income tax rate differential
|
( | ) | ( | ) | ||||
|
U.S. state income taxes, before valuation allowance
|
( | ) | ( | ) | ||||
|
Other
|
( | ) | ||||||
|
Change in valuation allowance
|
||||||||
|
Reported income tax expense
|
||||||||
The Company is an emerging growth company and is not currently subject to the disclosure requirements under ASU 2023-09. The Company has not adopted the standard as of the year ended December 31, 2025.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
7.
PLANT AND EQUIPMENT, NET
|
(in thousands)
|
Estimated
Useful Lives |
2025
$ |
2024
$ | ||||||
|
Plant and equipment
|
|
||||||||
|
Capital work in progress
|
|||||||||
|
Information technology equipment, under finance lease
|
|
||||||||
|
Less accumulated depreciation
|
( | ) | ( | ) | |||||
Depreciation expense of $1.5 million and $1.3 million was recognized in fiscal years 2025 and 2024, respectively.
8.
LEASES
The Group’s lease agreements include leases accounted for as operating leases and those accounted for as finance leases. This note provides information for leases where the Group is a lessee.
The Group leases laboratory facilities and offices through operating leases. These leases typically include lease options to renew the lease at which time the lease payments are renegotiated or adjusted to reflect pre-agreed charges or market rentals. Extension and termination options are included in a number of the property leases to allow for flexibility in terms of corporate growth and managing the assets used in the Group’s operations.
The Group leases IT equipment through finance leases with contract terms of 2 -3 years. In order to extend the leases, both parties must agree. Finance lease ROU assets are included in plant and equipment, net and finance lease liabilities are included in current debt obligations and long-term debt on the consolidated balance sheets. The ROU assets, lease liabilities, lease costs, cash flows, and lease maturities associated with the Group’s finance leases were not material to the consolidated financial statements for fiscal years 2025 and 2024.
The table below discloses the balance sheet information relating to the Group’s operating leases:
| (in thousands) | Balance sheet
Classification | 2025
$ | 2024
$ | ||||||
| ROU assets | |||||||||
| Current liability | Current Operating lease liabilities | ||||||||
| Non-current liability | Non-current Operating lease liabilities | ||||||||
Operating lease costs of $0.9 million and $0.8 million were recognized in fiscal years 2025 and 2024, respectively. The Company recognizes variable lease payments not included in its lease liabilities in the period in which the obligation for those payments is incurred. Short term and variable lease payments for fiscal year 2025 and 2024 were not material.
As of December 31, 2025, the weighted-average remaining lease terms of the Group’s operating leases was 3.8 years, compared to 2.6 years as of December 31, 2024. The increase primarily reflects the addition of the new 610 Maple Grove facility lease agreement to the Group’s operating lease portfolio during the year, following the transition from a subleased arrangement to a direct lease. The weighted-average discount rate applied to the Group’s operating leases was 11.4 % for the year ended December 31, 2025 and 14.8 % for the year ended December 31, 2024.
The following table summarizes the ROU assets obtained in exchange for lease liabilities and as a result of lease modifications:
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
ROU assets obtained in exchange for new operating lease liabilities
|
||||||||
|
Non-cash changes related to lease modifications, net of lease incentive
|
||||||||
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
8.
LEASES (continued)
The following table summarizes the maturities of the Company’s operating leases at December 31, 2025. The amounts disclosed in the table are the contractual undiscounted cash flows. It is not expected that the cash flows included in the below maturity analysis could occur significantly earlier, or at significantly different amounts.
|
Fiscal year
(in thousands)
|
Operating Leases
$ | |||
|
2026
|
||||
|
2027
|
||||
|
2028
|
||||
|
2029
|
||||
|
2030
|
||||
|
Total expected lease payments
|
||||
|
Less imputed interest
|
( | ) | ||
|
Total lease liabilities
|
||||
9.
OTHER ASSETS
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Current
|
||||||||
|
Holdback receivable (refer to Note 5)
|
||||||||
|
Research and development tax incentive receivable
|
||||||||
|
Deferred offering costs
|
||||||||
|
Lease incentive receivable
|
||||||||
|
Deferred insurance costs
|
||||||||
|
Advance to contract research organization
|
||||||||
|
Other receivables
|
||||||||
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
10.
ACCRUED AND OTHER LIABILITIES
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Current
|
||||||||
|
Accrued liabilities
|
||||||||
|
Employee compensation and withholdings
|
||||||||
|
Lease asset retirement obligation
|
||||||||
|
Estimated legal contingency liability
|
||||||||
|
Cash-settled stock-based payment provision
|
||||||||
|
Non-current
|
||||||||
|
Employee compensation and retirement benefits
|
||||||||
|
Lease asset retirement obligation
|
||||||||
|
Cash-settled stock-based payment provision
|
||||||||
|
Other variable liabilities
|
||||||||
Estimated legal contingency liability
In 2024, the Company recognized a $1.4 million provision in connection with a legal claim related to the Company’s equity raise and subsequent IPO. The matter was settled in 2025, and the liability was extinguished in full.
Cash-settled stock-based payment provision
Refer to Note 15 Stock-based Compensation.
Lease asset retirement obligation
The lease asset retirement obligation relates to the removing of leasehold improvements including laboratories, clean rooms and office spaces and returning the premises to their original condition in accordance with the lease agreements. The calculation of this obligation requires assumptions such as closure dates and cost estimates. The amount recognized for each site is periodically reviewed and updated based on the facts and circumstances available at the time. Changes to the estimated future costs for sites are recognized in the balance sheet by adjusting the asset and the liability.
The obligation is classified as current based on the contractual lease expiry in 2025.
11.
DEBT OBLIGATIONS
Convertible Note Facility
On October 31, 2024, ATPL entered into a secured convertible note facility (the “Convertible Note Facility”) with Obsidian Global Partners, LLC (“Obsidian”), which was assumed by the Company upon completion of the Scheme. Under the initial $5.0 million (A$7.5 million) drawdown, ATPL became obligated to issue 75,000 options or pay a fee of 3 % of the AUD drawdown amount.
On December 16, 2024, following the closing of the Company’s IPO and the Scheme, Obsidian exercised its right to redeem the aggregate outstanding convertible notes for cash. On December 19, 2024, the Company paid Obsidian $5.7 million for the aggregate outstanding convertible notes and an additional $0.2 million in lieu of the options required to be issued in connection with the first drawdown. The Company recognized a loss on debt extinguishment in connection with the redemption. No convertible notes were outstanding under the Convertible Note Facility on December 31, 2024, and on February 18, 2025, the facility was terminated and the related security interest was released.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
11.
DEBT OBLIGATIONS (continued)
Contractual obligations
The Company had no significant financing arrangements outstanding as of December 31, 2025, or December 31, 2024.
12.
FAIR VALUE MEASUREMENT
The consolidated financial statements include financial instruments for which the fair value of such instruments may differ from the amounts reflected on a historical cost basis. Financial instruments consist of cash deposits, accounts and other receivables, accounts payable, accrued liabilities and debt obligations. The carrying value of these financial instruments generally approximates fair value due to their short-term nature.
For the periods presented, the total balances of financial assets and liabilities measured or disclosed at fair value were not material to the consolidated financial statements. As such, management has concluded that detailed quantitative disclosures, including the fair value hierarchy table, are not necessary. The carrying amounts of the Company’s remaining financial instruments approximate their fair values.
13.
EQUITY
(a)
Share Capital
The Company’s authorized share capital consists of 400,000,000 shares of Common Stock, par value $0.0001 per share, and 40,000,000 shares of preferred stock, par value $0.0001 per share (“preferred stock”).
Stockholders of the Company hold either Common Stock or a CHESS Depositary Interest (“CDI”). CDIs confer the beneficial ownership of the Company’s Common Stock on each CDI holder, with the legal title to such securities held by an Australian depositary entity, CHESS Depositary Nominees Pty Limited (the “Depositary Nominee”), which is a wholly-owned subsidiary of ASX Limited, being the operator of the ASX. The Depositary Nominee will be the registered holder of those shares of our Common Stock held for the benefit of the holders of CDIs.
The Company has never declared or paid any cash dividends on its capital stock and does not anticipate paying any cash dividends in the foreseeable future.
(b)
Movements in Common Stock
During the year ended December 31, 2025, the Company issued shares of Common Stock in connection with the following transactions:
●
In January 2025, in connection with the Company’s IPO which closed on December 16, 2024, TD Cowen, Barclays and Cantor (in their capacity as the underwriters’ representatives in the IPO) partially exercised the over-allotment option granted by the Company, pursuant to which the Company issued and sold an additional 78,481 shares of Common Stock at the purchase price of $6.00 per share for incremental gross proceeds of $0.5 million.
●
In March 2025, certain directors exercised a total of 289,500 stock options, resulting in the issuance of an aggregate 32,959 shares of Common Stock. Of these:
-
A portion of the options were net exercised, with the intrinsic value of the awards used to settle the exercise price and taxes. Shares withheld to facilitate net settlement were accounted for as share repurchases.
-
A portion of the options were exercised for cash, generating total cash proceeds of $114,833 .
●
In March 2025, external investors exercised 10,000 stock options for $6.22 per share (A$10.00 ), for gross proceeds of $0.1 million.
●
Periodically through the year, 1,314 unlisted stock options were exercised by employees (excluding directors and named executive officers). These options had a weighted average exercise price of $3.43 per share.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
13.
EQUITY (continued)
●
In October and November 2025, the Company completed private placement offerings of equity securities to certain investors. The Company issued (i) 2,346,936 one share of Common Stock, at a price of $4.90 per share and warrant, and (ii) 2,788,064 one CDI, at a price of A$7.50 per CDI and warrant.
The U.S. Warrants and CDI Warrants (collectively the “Warrants”) were classified as equity instruments and were measured at fair value on the issuance date. The portion of the proceeds attributable to the Warrants has been recorded within additional paid‑in capital. The fair value allocated to the U.S. Warrants and CDI Warrants on issuance was $4.2 million and $5.1 million, respectively. These amounts represent the estimated fair value of the Warrants at the issuance dates and do not affect the par value of the underlying Common Stock. The Company will not recognize subsequent fair value changes for the Warrants given their equity classification.
The Common Stock and CDI private placements generated aggregate gross proceeds of approximately $25.2 million. The Common Stock offering closed on October 27, 2025, and the CDI offering closed on November 5, 2025. In connection with the transaction, the Company also issued 250,000 CDI warrants to the lead manager.
●
In December 2025, a total of 611,108 RSUs vested. Upon settlement, the Company issued 611,108 shares of Common Stock, of which 228,797 shares were withheld to satisfy employee payroll tax‑withholding obligations. The withheld shares were accounted for as share repurchases. Accordingly, the Company issued 382,311 shares upon settlement.
Subsequent to the end of the reporting period, the Company issued 55,652,173 shares of Common Stock. Refer to note 23 Subsequent Events for details.
During the year ended December 31, 2024 and prior to the reverse recapitalization, ATPL issued the following ordinary shares:
●
In January 2024, 667 unlisted stock options issued under the Equity Plan (as defined below) were exercised. These options had an exercise price of $5.67 equivalent per share (A$8.60 ).
●
At various dates throughout the year, external investors exercised 403,000 unlisted stock options for $6.58 equivalent per share (A$10.00 ) for gross proceeds of $2.7 million.
●
In April 2024, 1,000,000 new shares were issued to various sophisticated and professional investors at $14.74 equivalent per share (A$23.00 ) for gross proceeds of $14.7 million.
●
In July 2024, 1,875,000 new shares were issued to various sophisticated and professional investors at $10.49 equivalent per share (A$16.00 ) for gross proceeds of $19.7 million.
●
In July 2024, 41,000 new shares were issued to a consultant for services provided. The equivalent price per share was $10.49 (A$16.00 ).
In connection with the IPO and following completion of the reverse recapitalization in December 2024, ATGC issued 14,800,000 shares of Common Stock at a public offering price of $6.00 per share for gross proceeds of $88.8 million prior to underwriting discounts, commissions and other estimated offering expenses.
(c)
Equity-linked securities held by investors
As of December 31, 2025 and December 31, 2024, the Company had 9,942,398 and 6,797,806 equity-linked securities outstanding, respectively, consisting of warrants, stock options and RSUs, with varying exercise prices and expiration dates. Equity-linked securities issued as stock-based compensation are described in note 15 Stock-based Compensation. The remaining securities held by investors totaled 5,135,000 and 1,958,933 as of December 31, 2025 and 2024, respectively, all of which are exercisable for shares of Common Stock, with exercise prices ranging from $7.50 to $7.70 and expiration dates between October and November 2030.
As of December 31, 2025 and December 31, 2024, these securities had no 7.61 and $15.37 as of December 31, 2025 and December 31, 2024, respectively.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
14.
LOSS PER SHARE
The table below presents the computation of basic and diluted loss per share:
|
(in thousands)
|
2025
$ |
2024
$ | |||||||
|
Loss for the year, attributable to the owners of the Company
|
$’000 | ( | ) | ( | ) | ||||
|
Weighted average number of shares outstanding: used in the denominator in calculating basic and diluted loss per share
|
Number | ||||||||
|
Basic and diluted loss per share
|
$ | ( | ) | ( | ) | ||||
|
Securities excluded as their inclusion would be anti-dilutive
|
Number | ||||||||
The Company reported net losses for all periods presented. As a result, the inclusion of any potentially dilutive shares of Common Stock would have been anti‑dilutive. Accordingly, stock options, RSUs, and warrants to purchase an aggregate of 9,942,398 shares of Common Stock were excluded from the calculation of diluted earnings per share. These instruments may have a dilutive effect on earnings per share in future periods.
Subsequent to the end of the reporting period, the Company issued 55,652,173 shares of Common Stock. Refer to note 23 Subsequent Events for details. The issuance of the additional shares of Common Stock has a material impact on the Company’s loss per share calculations. After the issuance, the basic and diluted loss per share for the year ended December 31, 2025 would have been $0.97 1.58 per share.
The financial statements for the year ended December 31, 2025 do not reflect the effects of this issuance, which will be included in the financial statements for the subsequent period.
15.
STOCK-BASED COMPENSATION
The Company grants stock-based compensation to directors, executive officers, employees and other service providers under the ATGC Equity Incentive Plan (the “Equity Plan”) which was established in December 2024. Stock awards, including restricted stock, RSUs, cash incentive awards, performance shares, performance units (“PSUs”), and other equity-based awards may be granted under the Equity Plan. No further grants will be made under the incentive plans previously maintained by ATPL.
As of December 31, 2025, 2,610,662 shares of Common Stock remained available for issuance under the Equity Plan as incentive stock options (“ISO”), subject to the ISO sublimit. On January 1, 2026, the Equity Plan’s share reserve increased by an additional 2,576,113 shares pursuant to the Equity Plan’s evergreen provision.
Any references in the Equity Plan and this summary to “shares of Common Stock” may be read as a reference to a CDI or shares of Common Stock as the context reasonably requires.
The Company issues new shares upon the vesting of equity awards and permits net share settlement to satisfy employee tax withholding obligations. Shares withheld to satisfy tax obligations are accounted for as share repurchases.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
15.
STOCK-BASED COMPENSATION (continued)
(a)
Stock-based compensation expense
The following table presents the components and classification of stock-based compensation expense recognized for stock options, cash-settled stock-based payments rights (“SPP rights”), RSUs and shares of Common Stock issued to employees, directors and consultants:
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Equity-settled stock-based payments (including stock options and RSUs)
|
||||||||
|
Cash-settled stock-based payments (SPP rights)
|
( | ) | ||||||
|
Shares issued as compensation to consultants
|
||||||||
|
Total stock-based compensation expense
|
||||||||
|
Classification of stock-based compensation expense
|
||||||||
|
Cost of products sold
|
||||||||
|
Research and development expense
|
||||||||
|
Selling, general and administrative expense
|
||||||||
|
Total stock-based compensation expense
|
||||||||
|
Stock-based compensation capitalized to equity (transaction cost)
|
||||||||
|
Income tax benefit
|
||||||||
|
Total stock-based compensation
|
||||||||
As of December 31, 2025, there was $8.2 million of total unrecognized compensation cost related to non-vested stock-based compensation arrangements granted. That cost is expected to be recognized over a weighted-average period of 1.6 years.
(b)
Stock options
Stock options issued by the Company to employees, directors and consultants have been described below. Each stock option, when exercised, entitles the holder to subscribe for and be allotted one share of Common Stock in the Company. Stock options granted in fiscal year 2025 were under the Equity Plan.
In connection with the reverse recapitalization completed in December 2024, all outstanding ATPL equity awards were exchanged for equivalent awards in ATGC. The exchange did not change the fair value, vesting conditions, or other terms of the awards, and therefore did not result in modification accounting under ASC 718.
The key terms of the Company’s equity incentive plans, as they relate to stock options, include:
●
Stock options held by Australian employees have an AUD base currency and convert into CDIs. All other options held by non-Australian employees have a USD base currency and convert into shares of Common Stock;
●
The exercise prices of the stock options are determined by the Board in its absolute discretion. In prior periods, exercise prices were generally determined with reference to the 5 -day or 20 -day volume-weighted average price (“VWAP”) of the Company’s listed shares of Common Stock. The Company now uses the closing price of its Common Stock on the trading day immediately prior to the grant date;
●
Stock options vest in three equal tranches on the completion of at least 12 , 24 and 36 months of continued service;
●
Stock options expire 5 or 10 years after the grant date;
●
All stock options expire on the earlier of their contractual expiration date or 90 days following the individual’s termination of the employment;
●
Stock options are unlisted and not transferable unless the Board in its absolute discretion agrees to a transfer;
●
Stock options carry no dividend rights or voting rights; and
●
If a change of control event occurs prior to the vesting of an award, then the Board may determine, in its absolute discretion, the treatment of the participant’s unvested awards and the timing of such treatment.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
15.
STOCK-BASED COMPENSATION (continued)
Director stock options
In March 2025, all outstanding stock options with share price performance hurdles held by directors (289,500 options at a weighted-average exercise price of $7.12 ) were exercised, resulting in the issuance of 32,959 shares of Common Stock. Refer to note 13 Equity. No stock options with share price performance hurdles remained outstanding as of December 31, 2025.
In June 2024, ATPL issued 475,000 stock options to directors at an exercise price of $15.34 equivalent (A$23.00 ) per share. These options expire after 5 years and vest in three tranches on the completion of at least 12 , 24 and 36 months of continued service. In June 2025, 50,000 unvested options were forfeited upon a director’s resignation in accordance with the original terms of the awards.
Employee stock options
The number of employee stock options issued during the years ended December 31, 2025 and 2024 was 52,750 and 166,700 , respectively. These quantities exclude stock options issued to directors.
Consultant stock options and share grants
During the year ended December 31, 2025, all outstanding stock options issued to consultants under stock-based payment arrangements, representing 1,000,000 options at a weighted-average exercise price of $12.12 , expired unexercised. As a result, no consultant stock options were outstanding as of December 31, 2025.
Modification of Stock Options
In December 2025, the Company amended the terms of its outstanding stock options following approval by stockholders. Prior to the amendment, optionholders were required to pay the applicable exercise price in cash in order to exercise vested options and were generally responsible for any associated tax obligations at the time of exercise.
The amendment provides the Board with discretionary authority to (i) permit optionholders to exercise their vested options using a net exercise (cashless exercise) feature and (ii) allow net share withholding to satisfy the tax liabilities incurred upon exercise. Under the net exercise feature, the Company issues a reduced number of shares by withholding shares with a value equal to the applicable exercise price at exercise. Under the net share‑withholding feature, the Company may also withhold shares with a value equal to the employee’s tax obligation at exercise. All shares withheld in connection with these features are accounted for as share repurchases.
The amendments did not modify the vesting conditions, contractual term, or other substantive terms of the options. The modification did not change the classification of the awards and did not result in any incremental fair value, as the fair‑value‑based measure of the options immediately before and after the modification was substantially the same. Accordingly, no additional stock‑based compensation expense was recognized in connection with the modification.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
15.
STOCK-BASED COMPENSATION (continued)
Service-based stock options held by employees and directors
The number and weighted-average exercise prices of service-based stock options (employee and director options), excluding those with share price performance hurdles, under stock-based payment arrangements were as follows:
| Number of options | Weighted-
average exercise price $ | Weighted-
average Remaining Contractual Term (in years) | Aggregate
Intrinsic Value (in thousands) $ | |||||||||||||
| Outstanding at January 1, 2025 | ||||||||||||||||
| Granted during the year | ||||||||||||||||
| Forfeited during the year | ( | ) | ||||||||||||||
| Exercised during the year | ( | ) | ||||||||||||||
| Expired during the year | ( | ) | ||||||||||||||
| Outstanding at December 31, 2025 | ||||||||||||||||
| Expected to vest at December 31, 2025 | ||||||||||||||||
| Exercisable at December 31, 2025 | ||||||||||||||||
A change of control event is a non-market condition which has not been taken into consideration in the valuation of the options. A change of control event was not considered probable as of December 31, 2025.
Fair value of stock options
The Company uses the Black-Scholes model to determine the fair value of stock options at the grant date. Determining the fair value of these awards requires management to make estimates and assumptions, including projected employee stock option exercise behaviors, expected price volatility of the underlying share, the expected dividend yield and the risk-free interest rate over the expected term.
The following table presents the fair value of stock-based payments options granted to directors during the year and the inputs used in the Black-Scholes model.
| 2025 | 2024 | |||||||
|
Quantity issued during the period
|
||||||||
|
Weighted average fair value per stock option at grant date
|
$ | |||||||
|
Assumptions used:
|
||||||||
|
Share price at grant date
|
$ | |||||||
|
Exercise price
|
$ | |||||||
|
Expected volatility range
|
| |||||||
|
Expected life range
|
- | | ||||||
|
Expected dividends
|
| |||||||
|
Risk-free interest rate range
|
| |||||||
In connection with the Common Stock and CDI private placement that closed in late 2025, the lead manager received 250,000 CDI warrants as consideration for services rendered. The CDI warrants were measured at fair value on the grant date and accounted for as equity‑settled share‑based compensation, with the corresponding amount capitalized as a cost of the capital raise and recorded as a reduction of equity.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
15.
STOCK-BASED COMPENSATION (continued)
The following table provides the weighted average fair value of options granted to employees during the year and the related weighted average inputs (based on number of options granted) used in the Black-Scholes model.
| 2025 | 2024 | |||||||
|
Quantity issued during the period
|
||||||||
|
Weighted average fair value per stock option at grant date
|
$ | $ | ||||||
|
Assumptions used:
|
||||||||
|
Share price at grant date range
|
$ | $ | ||||||
|
Exercise price range
|
$ | $ | ||||||
|
Expected volatility range
|
| | ||||||
|
Expected life range
|
| | ||||||
|
Expected dividends
|
| | ||||||
|
Risk-free interest rate range
|
| | ||||||
The following table summarizes the status of Anteris’ non-vested service-based stock options:
| Number of options |
Weighted-average
exercise price $ | |||||||
|
Non-vested at December 31, 2024
|
||||||||
|
Granted
|
||||||||
|
Vested
|
( | ) | ||||||
|
Forfeited
|
( | ) | ||||||
|
Non-vested at December 31, 2025
|
||||||||
(c)
Share Price Performance Rights (Cash-settled)
The Share Price Performance Plan provides employees with the right to receive cash payments calculated by considering the rise in Anteris’ share price from the base price specified at grant date. The SPP rights expire after 5 years. There are two types of arrangements:
●
Service based conditions: employees have the right to receive cash payments after 1 , 2 and 3 years from the grant date subject to the holder still being employed by the Group. The cash payments are calculated by considering the rise in Anteris’ share price from the base price to the vesting date.
●
Service and performance conditions: the SPP rights are divided into three equal tranches which vest and become exercisable on the earlier of the achievement of specified share price hurdles and the completion of 3 years of service. The cash payments are calculated by considering the rise in Anteris’ share price from the base price to the exercise date.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
15.
STOCK-BASED COMPENSATION (continued)
The following table summarizes the SPP rights activity during the year:
| Number of rights | Weighted- average base price $ | Weighted- average Remaining Contractual Term (in years) | Carrying amount of liabilities (in thousands) $ | |||||||||||||
| SPP with service conditions | ||||||||||||||||
| Non-vested at January 1, 2025 | ||||||||||||||||
| Forfeited during the year | ( | ) | ||||||||||||||
| Non-vested at December 31, 2025 | ||||||||||||||||
| SPP with service and performance conditions | ||||||||||||||||
| Non-vested at January 1, 2025 | ||||||||||||||||
| Non-vested at December 31, 2025 | ||||||||||||||||
Fair value of SPP
The Company uses the Black-Scholes model to determine the fair value of SPP rights at the grant date. Because SPP rights are liability‑classified, their fair value is subsequently remeasured at each reporting date using the Black‑Scholes model.
The inputs used in the measurement of the fair values at reporting date of the SPP rights were as follows:
| Service based SPP | December 31, 2025 | December 31, 2024 | ||||||
| Weighted average fair value per right | $ | $ | ||||||
| Share price at measurement date | $ | $ | ||||||
| Base price | $ | $ | ||||||
| Expected volatility (weighted average) | % | % | ||||||
| Expected life (weighted average) | | | ||||||
| Risk-free interest rate (based on government bonds) | % | % | ||||||
| Service and performance based SPP | December 31, 2025 | December 31, 2024 | ||||||
| Weighted average fair value per right | $ | $ | ||||||
| Share price at measurement date | $ | $ | ||||||
| Base price | $ | $ | ||||||
| Expected volatility (weighted average) | % | % | ||||||
| Expected life (weighted average) | | | ||||||
| Risk-free interest rate (based on government bonds) | % | % | ||||||
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
15.
STOCK-BASED COMPENSATION (continued)
(d)
RSU
RSUs generally vest over 3 years, with compensation cost, adjusted for estimated forfeitures, being recognized on a straight-line basis over the requisite service period for each separately vesting portion of the award as if the award was, in-substance, multiple awards. Certain non‑employee directors, as defined by Rule 16b-3 promulgated under the Exchange Act, receive annual grants of RSUs that vest on the earlier of (i) the first anniversary of the grant date and (ii) the date of the next annual meeting of stockholders, subject to continued service through the vesting date. RSUs are not considered issued or outstanding Common Stock. Dividend equivalent units are accumulated on RSUs during the vesting period and are subject to the same restrictions on transferability as the underlying RSUs.
The following table summarizes the RSU activity during the year:
| Number of RSUs |
Weighted-average
grant price $ | |||||||
|
Non-vested at January 1, 2025
|
||||||||
|
Granted during the year
|
||||||||
|
Settled in shares during the year
|
( | ) | ||||||
|
Forfeited during the year
|
( | ) | ||||||
|
Non-vested at December 31, 2025
|
||||||||
Fair Value of RSUs
The fair value of the RSUs is based on the market value of the Common Stock on the date of grant.
The weighted-average grant date fair value of RSUs granted was $4.16 per RSU during the year ended December 31, 2025 and $6.00 per RSU during the year ended December 31, 2024. The fair value of the RSUs was determined based on the market price of the Common Stock on the grant date, which represents the fair value of the underlying shares.
During the year, the Company issued RSUs to directors that had been contingently granted in December 2024. Although the indicative fair value at that date was $6.00 per RSU based on the then‑current market price of the Common Stock, the awards were not recognized until stockholder approval was obtained, in accordance with ASC 718. Upon approval, the grant‑date fair value was measured at $4.46 per RSU, based on the market price of the Common Stock on the approval date, which represents the grant date for accounting purposes.
(e)
Other information
The following table summarizes the Company’s cash flows and other information related to stock‑based payment awards for the year.
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Cash proceeds from options exercised
|
||||||||
|
Cash paid for withholding taxes
|
( | ) | ||||||
|
Cash payments for stock-based liabilities
|
||||||||
|
Intrinsic value of options exercised
|
||||||||
|
Intrinsic value of restricted stock units vested
|
||||||||
|
Fair value of restricted stock units vested
|
||||||||
|
Income tax benefit related to options exercised
|
||||||||
In connection with the Common Stock and CDI private
placement that closed in late 2025, the lead manager received 250,000 CDI
warrants as consideration for services rendered. The CDI warrants were measured
at fair value on the grant date and accounted for as equity-settled share-based
compensation, with the corresponding amount capitalized as a reduction of
equity.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
16.
VARIABLE INTEREST ENTITY
The Company has agreed to provide certain development services to v2vmedtech in exchange for equity in v2vmedtech. We provide engineering, clinical, regulatory, marketing, and executive management resources, but excluding medical and chief medical officer services, in connection with v2vmedtech’s development of an innovative heart valve repair device utilizing a transcatheter edge-to-edge repair method for a minimally invasive treatment of mitral and tricuspid valve regurgitation, also known as leaky valve.
The Company has determined that v2vmedtech is a VIE under ASC 810 and that the Company is the primary beneficiary because it has the power to direct the activities that most significantly affect v2vmedtech’s economic performance, primarily through appointing and holding a majority of the v2vmedtech’s board of directors, and has the right to receive certain benefits or the obligation to absorb losses that could potentially be significant to v2vmedtech through equity ownership. Therefore, the Company consolidates v2vmedtech and reassesses its primary‑beneficiary status at each reporting date.
Our involvement with v2vmedtech affects our financial position through the inclusion of these assets and liabilities, and affects our cash flows through the consolidation of its operating cash flows. Because v2vmedtech is an early‑stage R&D medical technology company, our involvement exposes us to risks primarily related to funding ongoing research activities and absorbing losses from its operations. These risks have not materially changed during the period.
The Company provides non-reciprocal contributions to fund R&D.
The following table presents the assets and liabilities for VIE:
| As of December 31, | ||||||||
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
| Assets | ||||||||
| Other current assets | ||||||||
| Total assets | ||||||||
| Liabilities | ||||||||
| Other current liabilities | ||||||||
| Non-current liabilities | ||||||||
| Total liabilities | ||||||||
| Net (liabilities)/assets | ( | ) | ( | ) | ||||
Included in other current liabilities is a loan from the Company, with is v2vmedtech’s parent entity, amounting to $14,969 as of December 31, 2025, and $20,798 as of December 31, 2024. This loan has been provided to support v2vmedtech’s working capital needs. It is unsecured and repayable on demand. This balance is eliminated in the condensed consolidated financial statements. Other than the initial capital contributions of other stockholders of v2vmedtech, v2vmedtech is wholly financed by the Group. In exchange for v2vmedtech equity interests, the Group contributed $2.6 million and $2.4 million to v2vmedtech to finance its operations during the years ended December 31, 2025 and December 31, 2024, respectively.
Non-controlling Interests
Non-controlling interests represent the equity in a subsidiary not attributable, directly or indirectly, to the parent company. The Group uses the Hypothetical Liquidation at Book Value (“HLBV”) approach to measure the non-controlling interests. Under HLBV, the non-controlling interests are calculated as the amount that would be paid to non-controlling interest holders upon a hypothetical liquidation of the entity at book value as of the reporting date.
The Company recognizes non-controlling interests related to v2vmedtech and provides a roll forward of the non-controlling interests balance, as follows (in thousands):
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Opening balance at January 1
|
( | ) | ( | ) | ||||
|
Net (loss)/gain attributable to non-controlling interests
|
( | ) | ||||||
|
Closing balance at December 31
|
( | ) | ( | ) | ||||
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
17.
ACCUMULATED OTHER COMPREHENSIVE LOSS
The following table presents the components of Accumulated Other Comprehensive Loss:
|
(in thousands)
|
Foreign currency
translation adjustments $ |
Total Accumulated
Other Comprehensive Loss $ | ||||||
|
December 31, 2023
|
( | ) | ( | ) | ||||
|
Other comprehensive loss – equity adjustment from foreign currency translation
|
( | ) | ( | ) | ||||
|
December 31, 2024
|
( | ) | ( | ) | ||||
|
Other comprehensive income – equity adjustment from foreign currency translation
|
||||||||
|
December 31, 2025
|
( | ) | ( | ) | ||||
18.
RELATED PARTY TRANSACTIONS
Parent entities
The accounting parent entity of the Group is ATPL. The legal parent entity within the Group is ATGC.
Subsidiaries
The Company provides financial support to its subsidiaries from time to time when required, including letters of support.
The Group continues to consolidate v2vmedtech. The Group acquired a 30 % interest of v2vmedtech in 2023 and accounts for this interest as a variable interest entity for which the Company is the primary beneficiary (see Note 16 Variable Interest Entity).
There were no material changes in the Company’s ownership interests in subsidiaries during the year ended December 31, 2025.
19.
COMMITMENTS AND CONTINGENCIES
As of December 31, 2025 the Group had commitments to purchase $0.1 million of plant and equipment, as compared to $0.3 million at December 31, 2024.
Anteris is involved in various ongoing proceedings arising in the normal course of business, including proceedings related to product, labor, intellectual property and other matters.
Contingent liabilities
The Group records a liability in the consolidated financial statements on an undiscounted basis for loss contingencies related to legal actions when a loss is considered probable, and the amount may be reasonably estimated. If the reasonable estimate of a probable loss is a range, and no amount within the range is a better estimate than any other, the minimum amount of the range is accrued. If a loss is reasonably possible but not probable, and may be reasonably estimated, the estimated loss or range of loss is disclosed. When determining the estimated loss or range of loss, significant judgment is required. Estimates of probable losses resulting from litigation and governmental proceedings involving the Company are inherently difficult to predict, particularly when the matters are in early procedural stages with incomplete scientific facts or legal discovery, involve unsubstantiated or indeterminate claims for damages, potentially involve penalties, fines or punitive damages, or could result in a change in business practice.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
20.
SEGMENT REPORTING
(a)
Description of segments
Segment information is presented using a management approach, meaning that segment information is provided on the same basis as information is used for internal reporting purposes by the CODM which is the Vice Chairman and Chief Executive Officer, who makes key strategic decisions. The CODM is responsible for the allocation of resources and assessing the performance of the Group. Management has determined that the activities of the business as reviewed by the CODM are one segment, being the development and commercialization of the ADAPT® anti-calcification tissue. This is focused on the DurAVR® THV System.
(b)
Segment information
The revenue and cost information relating to all of the ADAPT® products including both the DurAVR® THV System and regenerative tissue products are regularly reviewed by the CODM on an aggregate basis.
The CODM assesses performance and allocates resources based on the Company’s consolidated statements of operations and key components and processes of the Company’s operations are managed centrally. Segment asset information is not used by the CODM to allocate resources. As a single reportable segment entity, the Company’s segment performance measure is net income/(loss).
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Net sales from external customers
|
||||||||
|
Depreciation & amortization
|
( | ) | ( | ) | ||||
|
Interest income
|
||||||||
|
Interest expense
|
( | ) | ( | ) | ||||
|
Other segment items
|
( | ) | ( | ) | ||||
|
Segment net loss
|
( | ) | ( | ) | ||||
Other segment items include operating expenses and income and expense items that are not separately disclosed and are included in the measure of segment net loss. These amounts primarily consist of cost of revenues, selling, general and administrative expenses, stock‑based compensation, and other income and expense items.
The Company’s segment net loss is the same as its consolidated net loss, and there are no reconciling items between segment results and consolidated results.
No detailed asset information by reportable segment has been reported given that the single segment’s information is already presented in the consolidated balance sheets. Refer to the consolidated statements of cash flows for significant non-cash items and total expenditure for additions of long-lived assets.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
20.
SEGMENT REPORTING (continued)
(c)
Geographic information
Segment revenues (net sales) have been based on the geographic location of the customers taking possession of the products. Geographic long-lived assets are attributed to the country based on the physical location of the assets.
| Net sales | Long-lived assets, net | |||||||||||||||
|
(in thousands)
|
2025
$ |
2024
$ |
2025
$ |
2024
$ | ||||||||||||
|
United States
|
||||||||||||||||
|
Germany
|
||||||||||||||||
|
Australia
|
||||||||||||||||
|
Switzerland
|
||||||||||||||||
|
Sweden
|
||||||||||||||||
(d)
Major customers
The following table summarizes revenues from major customers that individually accounted for 10% or more of the Company’s total revenues during the years ended December 31, 2025 and 2024:
|
(in thousands)
|
2025
$ |
2024
$ | ||||||
|
Customer A
|
||||||||
|
Customer B
|
||||||||
Amounts outstanding from these customers at reporting date were less than $0.1 million as of December 31, 2025, and $0.2 million as of December 31, 2024.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
21.
VALUATION AND QUALIFYING ACCOUNTS
| Additions | Deductions | |||||||||||||||||||||||||||
| Description (in thousands) | Balance at beginning of period $ | Charged to Costs and Expenses $ | Charged to Other Accounts $ | Charged to Costs and expenses $ | to Other Accounts $ | Balance at
End of Period $ | Net change $ | |||||||||||||||||||||
| Allowance for doubtful accounts | ||||||||||||||||||||||||||||
| Year ended December 31, 2025 | - | - | - | - | - | - | | |||||||||||||||||||||
| Year ended December 31, 2024 | - | - | - | - | - | - | | |||||||||||||||||||||
| Inventory reserve | ||||||||||||||||||||||||||||
| Year ended December 31, 2025 | - | - | - | - | - | - | | |||||||||||||||||||||
| Year ended December 31, 2024 | - | - | - | - | - | - | | |||||||||||||||||||||
| Deferred tax asset valuation allowance | ||||||||||||||||||||||||||||
| Year ended December 31, 2025 | ( | ) | | |||||||||||||||||||||||||
| Year ended December 31, 2024 | | ( | ) | ( | ) | |||||||||||||||||||||||
Allowance for doubtful accounts
The allowances for doubtful accounts deductions represent accounts receivable which have been written off. The Group’s revenues are primarily derived from two external customers, both of which have no recent history of default. As of December 31, 2025 and 2024, no
Inventory reserve
When applicable, the Group maintains reserves for excess or slow-moving inventory, and inventory which is obsolete, damaged, nearing its expiration date, or slow moving. Estimates are made regarding the future recoverability of the costs of these products and record provisions based on historical experience, expiration of sterilization dates and expected future trends. At December 31, 2025 and 2024, it was determined that no
Deferred tax asset valuation allowance
The deferred tax asset valuation allowances are provided for all deferred tax assets that are not be recognized due to insufficient future taxable income. Amounts charged to other accounts includes valuation allowance movements which are recorded in other comprehensive income as a part of foreign currency translation adjustments.
22.
DEED OF CROSS GUARANTEE
ATGC and ATPL (ACN 088 221 078) are party to a deed of cross guarantee dated December 20, 2024 (“Deed”). The Deed remained in place throughout the year ended December 31, 2025, and there were no changes to the parties to the Deed during that period. The Company did not rely on the reporting
relief available under the Deed for the year ended December 31, 2025. The Deed was entered into in prior periods for Australian statutory reporting purposes; however, the Company did not rely on the reporting relief available under the Deed for the year ended December 31, 2025, and the Deed had no impact on the preparation or presentation of these consolidated financial statements.
ANTERIS TECHNOLOGIES GLOBAL CORP.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEAR ENDED DECEMBER 31, 2025
23.
SUBSEQUENT EVENTS
Management has evaluated the impact of subsequent events through to February 25, 2026.
2026 Public Offering
On January 22, 2026, the Company completed an underwritten public offering (the “2026 Public Offering”) of 40,000,000 shares of its Common Stock, which included the full exercise of the underwriters’ option to purchase additional shares, at a public offering price of $5.75 per share. The 2026 Public Offering generated gross proceeds of approximately $230.0 million, prior to deducting underwriting discounts and commissions and estimated offering expenses.
The 2026 Public Offering was made pursuant to the Company’s shelf registration statement on Form S-3 (Registration No. 333-292565), which was previously filed with the Securities and Exchange Commission (the “SEC”) and declared effective on January 8, 2026, and a prospectus supplement dated January 20, 2026.
Medtronic Private Placement
On January 20, 2026, the Company entered into a stock purchase agreement with Covidien Group S.à r.l. (“Covidien”), a wholly owned subsidiary of Medtronic plc (together with Covidien, “Medtronic”), pursuant to which the Company issued and sold to Medtronic 15,652,173 shares of Common Stock at a purchase price of $5.75 per share (the “Medtronic Private Placement”). The Medtronic Private Placement closed on January 22, 2026, immediately after the completion of the 2026 Public Offering, and generated gross proceeds of approximately $90.0 million, before deducting placement agent fees and estimated offering expenses.
The issuance and sale of the shares of Common Stock to Medtronic in the Medtronic Private Placement was not registered under the Securities Act and were issued and sold in reliance on the exemption provided by Section 4(a)(2) of the Securities Act.
None.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation and supervision of our Chief Executive Officer and our Chief Financial Officer, have evaluated our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of December 31, 2025. Disclosure controls and procedures are designed to ensure that information we are required to disclose in reports we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that, as of December 31, 2025, our disclosure controls and procedures were not effective based on the material weaknesses discussed in Management’s Report on Internal Control over Financial Reporting described below.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with US GAAP. Internal control over financial reporting includes those policies and procedures which (a) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of assets, (b) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with US GAAP and that receipts and expenditures of the issuer are being made only in accordance with appropriate authorization of management and the directors of the issuer, and (c) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of assets that could have a material effect on the financial statements.
In connection with the preparation of our financial statements for the years ended December 31, 2024 and 2023, our management and our independent auditors identified material weaknesses in the design and operating effectiveness of our internal control over financial reporting, which remained unremediated as of December 31, 2025. The material weaknesses identified by our management and our independent auditors relate to (i) a lack of appropriately designed, implemented and documented procedures and controls, and (ii) deficiencies in the segregation of duties.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis.
Our management, with the participation and supervision of our Chief Executive Officer and our Chief Financial Officer, under the oversight of our Board, conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2025, based on the criteria set forth in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this evaluation, management concluded that our internal control over financial reporting was not effective as of December 31, 2025, as a result of the material weaknesses described above.
Remediation of Previously Reported Material Weakness
Management has initiated a remediation plan to address the material weaknesses described above. This plan includes formally documenting policies, processes, risks, and controls, and reviewing the design and operating effectiveness of key and non-key controls. Testing of operating effectiveness has commenced, and remediation actions are progressing as planned. Segregation of duties has been enhanced across the control environment and financial reporting systems through system automation, strengthened month-end controls, and strengthened month-end review procedures.
During the year ended December 31, 2025, we implemented changes to our internal control over financial reporting to remediate the material weaknesses disclosed in our prior filings. These remediation activities included enhancements to our control environment, improvements to process-level controls, and updates to documentation and oversight procedures. Management is currently validating and testing the operating effectiveness of these enhanced controls.
While we believe that these efforts will improve our internal control over financial reporting, the design and implementation of our remediation is ongoing and will require validation and testing of the design and operating effectiveness of our internal controls over a sustained period of financial reporting cycles. The actions that we are taking are subject to ongoing senior management review, as well as oversight by the Audit and Risk Committee. We will not be able to conclude whether the steps we are taking will fully remediate the material weaknesses in our internal control over financial reporting until we have completed our remediation efforts and subsequent evaluation of their effectiveness.
Changes in Internal Control over Financial Reporting
Other than the ongoing remediation plans described above, there were no changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the fourth quarter ended December 31, 2025, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Attestation Report of Registered Public Accounting Firm
This Form 10-K does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting due to an exemption established by the JOBS Act for EGCs.
Inherent Limitation on the Effectiveness of Internal Controls and Procedures
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the controls. The design of any system of controls is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
Insider Trading Arrangements and Policies
During the fiscal quarter ended December 31, 2025, none of our directors or officers (as defined in Section 16 of the Exchange Act) adopted terminated
None.
Board of Directors
Our Board oversees the management of the business and affairs of the Company and serves as the ultimate decision-making body of the Company, except for those matters reserved to our stockholders. Our Board oversees the Company’s management team, to whom it has delegated responsibility for the Company’s day-to-day operations. While our Board’s oversight role is broad and may concentrate on different areas from time to time, its primary areas of focus are strategy, oversight, governance and compliance, as well as assessing management.
Our Charter and Bylaws provide for a classified Board. There are three classes of directors, with each class of directors serving three-year terms that end in successive years. We currently have authorized five directors. Our Charter and Bylaws do not limit the number of terms a member may be re-elected as a director. The following table provides the names, ages and positions of our directors as of February 25, 2026.
| Name | Current Position |
Independent Under Nasdaq Rules |
Age | |||||
|
Class II Directors (Terms Expiring at the 2026 Annual Meeting of Stockholders)
|
||||||||
|
David St Denis
|
President, Director | No | 57 | |||||
|
Class III Directors (Terms Expiring at the 2027 Annual Meeting of Stockholders)
|
||||||||
|
Wayne Paterson
|
Vice Chairman and Chief Executive Officer | No | 59 | |||||
|
David Roberts
(1)(2)(3)
|
Director | Yes | 61 | |||||
|
Class I Director (Term Expiring at the 2028 Annual Meeting of Stockholders)
|
||||||||
|
John Seaberg
(1)(2)(3)
|
Chairman of the Board of Directors | Yes | 74 | |||||
|
Gregory Moss
(1)(2)(3)
|
Director | Yes | 42 |
(1)
Member of the Audit and Risk Committee.
(2)
Member of the Compensation Committee.
(3)
Member of the Nominating and Corporate Governance Committee.
Our directors bring a range of skills and experience in relevant areas, including finance strategy, marketing, Biomedical Technology, global health care, public company director experience, corporate finance and capital investment and global/international corporate experience. We believe this cross-section of capabilities enables our Board to help guide our objectives and leading corporate governance practices. More detailed biographical descriptions of each director is set forth below.
John Seaberg
John Seaberg has been Chairman of our Board since March 2017 and a director since October 2014, in each case taking into account positions held with ATPL. Additionally, Mr. Seaberg has been serving as Chair of the board of directors of Preceptis Medical Inc since 2016 and Phraxis Medical Inc since 2009. He was Executive VP at Cedar Point Capital, a broker-dealer focused on healthcare investment, from June 2015 through December 31, 2023. From 2008 until 2012, Mr. Seaberg was Chair of Synovis Inc., a Nasdaq-listed manufacturer of various medical device and bioscaffold tissue products which was acquired by Baxter, and, from 2007 until 2014, was Co-Founder, Chair and Chief Executive Officer of NeoChord Inc., a company commercializing technology developed at the Mayo Clinic for repair of the mitral valve via minimally invasive techniques. From 1996 to 2006, Mr. Seaberg served at Guidant Corp. (subsequently acquired by Boston Scientific Corp.) where he held various executive level positions, including Director of Marketing for Cardiac Rhythm Management, Vice President of Sales for Cardiac Surgery and Vice President of Sales for Cardiac Rhythm Management. In addition, Mr. Seaberg was co-Founder, President and Chief Executive Officer of ACIST Medical, from 1991 to 1995. Mr. Seaberg holds a Bachelor of Arts in Speech Communications from the University of Minnesota and an MBA from the Carlson School of Management, also at the University of Minnesota. We believe that Mr. Seaberg’s qualifications to serve as a director include his extensive finance, leadership and industry experience and his tenure as a director of the Company.
Gregory Moss
Gregory Moss has served on our Board since June 2025. Mr. Moss serves as Chief Business and Legal Officer, as well as Corporate Secretary and Chief Compliance Officer of Evommune, Inc., a clinical stage biotechnology company dedicated to chronic inflammatory diseases. Prior to Evommune, Greg served as Executive Vice President, General Counsel, and Corporate Secretary, Chief Compliance Officer at Kadmon Corporation (“Kadmon”), a biopharmaceutical company, where he led legal, compliance, and business development operations, culminating in Kadmon’s acquisition by Sanofi in 2021. Prior to joining Kadmon in 2012, Mr. Moss served as a solicitor in the corporate risk department of a large Australian law firm and as an associate at a boutique law firm and hedge fund in New York, where he focused on complex litigation and event-driven outcomes. Mr. Moss currently serves on the board of directors of Vitls, Inc. Mr. Moss earned a BA and an LLB from Macquarie University, Australia, and is a member of the Bar Associations of New York, and New South Wales, Australia, with admissions before the Supreme Court of the United States of America; Southern District of New York; Supreme Court of New South Wales, Australia; and High Court of Australia. We believe that Mr. Moss’ qualifications to serve as a director include his extensive legal, leadership and industry experience.
David St Denis
David St Denis has served as our President and as a director since March 2025. Prior to that, Mr. St Denis served as the Company’s Chief Operating Officer since July 2017, taking into account his position with ATPL. In addition, Mr. St Denis has served as the Chief Executive Officer of v2vmedtech, which we consider to be a VIE and consolidate in our financial statements, since April 2023. Mr. St Denis also served as Chief Financial Officer of v2vmedtech from April 2023 to September 2023. Prior to his appointment as Chief Operating Officer of the Company, Mr. St Denis served as Head of Commercial Operations for Europe and Canada at Merck KGaA (“Merck”), a multinational science and technology company, since 2013, and prior to that, served as Head of Operations for Emerging Markets at Merck since 2008. In addition, Mr. St Denis had held multiple leadership roles at Millennium Pharmaceuticals, Inc, now Takeda Pharmaceutical Company, from 1996 to 2006, and provided strategic consulting services to such company from 2006 to 2008. Mr. St Denis has a Bachelor of Science from the University of Connecticut, a Master of Arts from Boston University and an MBA in Global Management and International Marketing from Babson College - Franklin W. Olin Graduate School of Business. We believe that Mr. St Denis’ qualifications to serve as a director include his business experience within the industry in which the Company operates, including as the Company’s President.
Wayne Paterson
Wayne Paterson joined the Company in October 2014 as a Non-Employee Director (as defined below), and has served as the Chair of the Board from February 2016 to March 2017, the interim Chief Executive Officer commencing in May 2016, and the Chief Executive Officer since March 2017, in each case taking into account positions held with ATPL, and has served as the Vice Chairman of our Board since March 2025. Mr. Paterson commenced service as Chair of the board of directors of v2vmedtech, which we consider to be a VIE and consolidate in our financial statements, in April 2023. Prior to joining the Company, Mr. Paterson held senior positions at Merck from 2005 to 2013, including as President of Europe, Canada and Australia, President of Emerging Markets, President of Japan and President of Cardiovascular Medicine. From 1999 until 2005, Mr. Paterson served at Roche Pharmaceuticals, a multinational healthcare company, in several senior positions, including as Head of Pharmaceuticals in Roche’s South Korean operation and Head of Commercial Operations for Roche China. Mr. Paterson holds an MBA from the University of Southern Queensland and a degree in Business Studies from the Queensland University of Technology. Mr. Paterson previously served as a director of Cepheid Inc. (NASDAQ: CHPD) from April 2015 to November 2016. We believe Mr. Paterson’s qualifications to serve as a director include his service as our Vice Chairman and Chief Executive Officer, as his insight into the business and related risks and challenges facing the Company will contribute to our Board and in its understanding of our business and strategy.
David Roberts
David Roberts has served on our Board since June 2025. Mr. Roberts is currently President of LeMaitre Vascular, Inc. (NASDAQ: LMAT), a provider of devices, implants, and services for the treatment of peripheral vascular disease, which is a position he has held since 2007. He joined LeMaitre in 1997 as Vice President of Business Development and was promoted to Chief Financial Officer in 2000. Prior to LeMaitre, Mr. Roberts served as Vice President of Development for BUCA, Inc. from 1994 to 1997, and prior to that as Associate of HarbourVest Partners from 1992 to 1994. Mr. Roberts received a Bachelor of Arts in Business Economics and History from Brown University and a Master of Business Administration from the Stanford University Graduate School of Business. Mr. Roberts serves as a director of LeMaitre Vascular, Inc., Lexington Medical, Inc. and Parasole Restaurant Holdings, Inc. We believe that Mr. Roberts’ qualifications to serve as a director include his extensive finance, leadership and industry experience.
Board Committees
Our Board has three standing committees: the Audit and Risk Committee, the Compensation Committee and the Nominating and Corporate Governance Committee. Each committee is governed by a charter that is available on our website at https://anteristech.com/investors/corporate-governance.html.
Audit and Risk Committee
The members of our Audit and Risk Committee consist of Mr. Seaberg, Mr. Roberts and Mr. Moss. Mr. Roberts is the chairperson of our Audit and Risk Committee. All three members of our Audit and Risk Committee meet the requirements for independence under the current Nasdaq Marketplace Rules (the “Nasdaq Listing Rules”) and Rule 10A-3 of the Exchange Act. Each member of our Audit and Risk Committee is “financially literate” under the Nasdaq Listing Rules. In addition, our Board has determined that Mr. Roberts is an “audit committee financial expert” within the meaning of the rules and regulations of the SEC. This designation does not impose on such director any duties, obligations, or liabilities that are greater than are generally imposed on members of our Audit and Risk Committee and our Board. Our Audit and Risk Committee is directly responsible for, among other things:
•
appointing, retaining, compensating and overseeing the work of our independent registered public accounting firm;
•
assessing the independence and performance of the independent registered public accounting firm;
•
reviewing with our independent registered public accounting firm the scope and results of the firm’s annual audit of our financial statements;
•
overseeing the financial reporting process and discussing with management and our independent registered public accounting firm the financial statements that we will file with the SEC;
•
pre-approving all audit and permissible non-audit services to be performed by our independent registered public accounting firm;
•
reviewing policies and practices related to risk assessment and management;
•
reviewing our accounting and financial reporting policies and practices and accounting controls, as well as compliance with legal and regulatory requirements;
•
reviewing, overseeing, approving, or disapproving any related-person and related-party transactions;
•
reviewing with our management the scope and results of management’s evaluation of our disclosure controls and procedures and management’s assessment of our internal control over financial reporting, including the related certifications to be included in the periodic reports we will file with the SEC;
•
establishing procedures for the confidential anonymous submission of concerns regarding questionable accounting, internal controls, or auditing matters, or other ethics or compliance issues;
•
reviewing reports from our management and our independent registered public accounting firm on the effectiveness of internal control, risk management systems and management of material business risks;
•
establishing and reviewing our risk management framework, including the risk profile developed by our management covering material risks to our business;
•
reviewing and assessing the effectiveness of our internal controls, policies, programs, guidelines and procedures making up our risk management framework and reporting systems, including in light of any material breakdowns and reports from our management on new or emerging sources of risk; and
•
reviewing with our management and recommending to our Board additional or material amendments to our risk management reporting and governance policies.
Compensation Committee
The members of our Compensation Committee consist of Mr. Seaberg, Mr. Moss and Mr. Roberts. Mr. Seaberg is the chairperson of our Compensation Committee. Each of Mr. Seaberg, Mr. Moss and Mr. Roberts is a non-employee director, as defined by Rule 16b-3 promulgated under the Exchange Act, and meets the requirements for independence under the current Nasdaq Listing Rules. Our Compensation Committee is responsible for, among other things:
•
reviewing and approving the compensation of our executive officers, including reviewing and approving corporate goals and objectives with respect to compensation;
•
administering our equity incentive plans;
•
reviewing and approving, or making recommendations to our Board with respect to, incentive compensation and equity plans;
•
reviewing and recommending that our Board approve compensation for our non-employee board members; and
•
establishing and reviewing general policies relating to compensation and benefits for our employees.
Nominating and Corporate Governance Committee
The members of our Nominating and Corporate Governance Committee consist of Mr. Seaberg, Mr. Moss and Mr. Roberts. Mr. Seaberg is the chairperson of our Nominating and Corporate Governance Committee. Mr. Seaberg, Mr. Moss and Mr. Roberts meet the requirements for independence under the current Nasdaq Listing Rules. Our Nominating and Corporate Governance Committee is responsible for, among other things:
•
identifying and recommending candidates for membership on our Board, including the consideration of nominees submitted by stockholders, and on each of our Boards’ committees;
•
reviewing and recommending our corporate governance guidelines and policies;
•
reviewing proposed waivers of the Code of Business Conduct and Ethics for directors and executive officers;
•
overseeing the process of evaluating the performance of our Board; and
•
assisting our Board on corporate governance matters.
Process for Nominating Potential Director Candidates
The Nominating and Corporate Governance Committee is responsible for identifying, screening and interviewing individuals qualified to become members of our Board and recommending qualified candidates for election by the stockholders consistent with the criteria set forth in the Company’s Corporate Governance Guidelines. In recommending nominees for director, the Nominating and Corporate Governance Committee will consider candidates who have a high level of personal and professional integrity, strong ethics and values and the ability to make mature business judgments. Additional criteria that the Nominating and Corporate Governance Committee and our Board may consider are:
•
the candidate’s experience in corporate management, such as serving as an officer or former officer of a publicly held company;
•
the candidate’s experience as a board member of another publicly held company, including those dual-listed in the United States and Australia;
•
the candidate’s professional and academic experience relevant to the Company’s industry;
•
the strength of the candidate’s leadership skills;
•
the candidate’s experience in finance and accounting and/or executive compensation practices;
•
whether the candidate has the time required for preparation, participation and attendance at Board meetings and committee meetings, if applicable; and
•
the viewpoints, background, experience and other characteristics.
The Nominating and Corporate Governance Committee considers all candidates equally. The Nominating and Corporate Governance Committee reviews the background and qualifications of each nominee to ensure that our Board has the requisite expertise and that its membership consists of persons with sufficiently diverse and independent backgrounds (taking into account the enhanced independence, financial literacy and financial expertise standards that may be required under law, Nasdaq rules or the 4th Edition of the ASX Corporate Governance Council’s Corporate Governance Principles and Recommendations (“ASX Operating Rules”).
The Nominating and Corporate Governance Committee values the input of stockholders in identifying director candidates. Accordingly, although the Nominating and Corporate Governance Committee does not have a specific policy with regard to the consideration of candidates recommended by stockholders, the Nominating and Corporate Governance Committee considers recommendations for Board candidates submitted by stockholders using substantially the same criteria as it applies to recommendations from the Nominating and Corporate Governance Committee, directors and members of management. Any such nominations should be submitted to the Nominating and Corporate Governance Committee in compliance with the procedural requirements set forth in our Bylaws.
Board Leadership Structure
The current Chairman of our Board is John Seaberg, who is an independent director under the Nasdaq Listing Rules and for the purposes of the ASX Operating Rules. The roles of Chairman of our Board and Chief Executive Officer are separate. Our Board believes that the separation of the offices of the Chairman of the Board and Chief Executive Officer allows our Chief Executive Officer to focus primarily on our business strategy, operations, and corporate vision.
Risk Oversight
While management is responsible for assessing and managing risks to the Company on a day-to-day basis, our Board oversees management’s efforts to assess and manage risk. Our Board (in conjunction particularly with the Audit and Risk Committee) monitors and receives advice on areas of operational and financial risk and considers strategies for appropriate risk management arrangements. Specific areas of risk which are periodically considered at meetings of our Board include financial management, information technology, people and culture, intellectual property, regulatory (medical), legal, manufacturing, legal and organizational transformation. Additional areas of focus for our Board include, but are not limited to:
•
managing the Company’s long-term growth;
•
strategic and operational planning, including significant acquisitions and the evaluation of the Company’s capital structure; and
•
legal and regulatory compliance.
More broadly, risks are considered in virtually every business decision and process and as part of the Company’s overall business strategy. While our Board has the ultimate oversight responsibility for the Company’s risk management policies and processes, the committees of our Board also have responsibility for risk oversight. As noted above, the Company’s Audit and Risk Committee assists our Board in overseeing our risk assessment and management processes related to, among other things, our financial reports and record-keeping, financial risk exposures, and cybersecurity risk, and the steps management has taken to monitor and control such exposures. The Compensation Committee is responsible for overseeing the management of risks relating to our compensation policies and practices, including policies providing for the recovery of incentive or equity-based compensation and limiting hedging activities related to Company stock. The Nominating and Corporate Governance Committee is responsible for risk oversight associated with corporate governance practices, the composition of our Board and its committees, and environmental, social and governance matters.
Our Board stays informed of each committee’s risk oversight and other activities via regular reports of the committee chairs to the full Board. Our Board’s role in risk oversight is consistent with the Company’s leadership structure, with the Chief Executive Officer and other members of senior management having responsibility for assessing and managing the Company’s risk exposure, and our Board and committees providing oversight in connection with those efforts.
Board and Committee Meetings and Attendance
Directors are expected to make every effort to attend all meetings of our Board and all meetings of the committees on which they serve. During fiscal year ended December 31, 2025, our Board had 8 Board meetings, our Audit and Risk Committee had 4 meetings, our Nominating and Corporate Governance Committee had no meetings and our Compensation Committee had 5 meetings. During such period, each member of our Board attended at 100% of all Board and relevant committee meetings held during the period in which such director served.
Additional Board Information
Each committee is at all times authorized under its charter to have direct, independent and confidential access to our other directors, management and personnel to carry out the committee’s purposes. Each committee is authorized to conduct or authorize investigations into any matters relating to the purposes, duties or responsibilities of the committee.
Each committee may, in its sole discretion, retain or obtain the advice of legal counsel, compensation or other consultants and other advisers. We must provide for appropriate funding, as determined by each committee, for payment of reasonable compensation to any legal counsel, compensation or other consultant or other adviser retained by the committee.
Executive Officers
The following table sets forth as of February 25, 2026, the names and ages of our executive officers, who also constitute our NEOs for 2025. Biographies for each executive officer that is not a director are included below the table. There are no family relationships between the executive officers or between any director and any executive officer.
|
Name
|
Age |
Position
|
||
|
Wayne Paterson
|
59 |
Vice Chairman and Chief Executive Officer
|
||
|
David St Denis
|
57 |
President, Director (former Chief Operating Officer)
|
||
|
Matthew McDonnell
|
53 |
Chief Financial Officer
|
Matthew McDonnell
Matthew McDonnell served as ATPL’s Chief Financial Officer from November 2018 and, following the Reorganization, now holds the role of Chief Financial Officer of the Company. He is also the Chief Financial Officer of v2vmedtech, which we consider to be a VIE and consolidate in our financial statements, since September 2023. Prior to his appointment as Chief Financial Officer of ATPL, Mr. McDonnell worked for KPMG, a global professional services firm, for over 24 years, where he held several senior positions, including 10 years as a partner. He has a broad range of industry experience and corporate governance acumen, having delivered audit, accounting, and advisory services to a broad range of sectors. During his time at KPMG, Mr. McDonnell worked in Australia covering the financial services, transport, industrial markets, health, childcare and energy industries. He has experience in restructurings, acquisitions, divestments, privatizations and other significant financial transactions. Mr. McDonnell also served as a director of the State Library of Queensland where he was the Chair of the Audit and Risk Management Committee for eight years. Mr. McDonnell holds a Bachelor of Economics from Macquarie University, Australia. He is an Associate of Chartered Accountants in Australia and New Zealand, a Fellow of the Financial Services Institute of Australasia and a Member of the Australian Institute of Company Directors.
Corporate Governance Matters
Our Board believes sound corporate governance processes and practices, as well as high ethical standards, are critical to handling challenges and to achieving business success. We embrace leading governance practices and also conduct ongoing reviews of our governance structure and processes to reflect stockholder input and changing circumstances. Below are highlights of our corporate governance practices and principles.
Our Board has adopted Corporate Governance Guidelines that outline our corporate governance policies and practices, which is available on our website at https://anteristech.com/investors/corporate-governance.html.
Code of Business Conduct and Ethics
We have adopted a written Code of Business Conduct and Ethics, which applies to all our directors, officers and employees, and is available on our website at https://anteristech.com/investors/corporate-governance.html.
The Audit and Risk Committee is responsible for overseeing the Code of Business Conduct and Ethics and must approve any waivers of the Code of Business Conduct and Ethics for executive officers and directors. We expect that any amendments to the Code of Business Conduct and Ethics, or any waivers of its requirements with respect to our executive officers and directors, will be disclosed on our website.
Compensation Committee Interlocks and Insider Participation
During the fiscal year ended December 31, 2025, our Non-Employee Directors (as defined below), Messrs. Seaberg, Denaro, Roberts and Moss, each served as a member of the Compensation Committee. Mr. Denaro served on the Compensation Committee until his resignation from the Board in December 2025, and Mr. Moss was appointed to the Compensation Committee following his appointment to the Board in June 2025. Other than Mr. Denaro, no member of the Compensation Committee has served as one of our officers or employees at any time. None of our executive officers currently serve, or in the past fiscal year has served, as a member of our Board and on the compensation committee of any other entity that has one or more of its executive officers serving on our Board or Compensation Committee.
Insider Trading and Securities Dealing Policy
We have adopted an Insider Trading and Securities Dealing Policy, which promotes compliance with insider trading laws, rules and regulations and the Nasdaq Listing Rules. Additionally, the policy prohibits officers, directors, and employees from engaging in short sales, publicly-traded options, or hedging transactions such as prepaid variable forwards, equity swaps, collars and exchange funds or through other transactions that hedge or offset, or are designed to hedge or offset, any decrease in the market value of our securities. Our officers, directors, and employees are also prohibited from including our securities in a margin account or pledging our securities as collateral for a loan.
Stockholder Communications
Any stockholder or other interested party who wishes to communicate with our Board or any individual director may send written communications to our Board or such director, care of Anteris Technologies Global Corp., Toowong Tower, Level 3, Suite 302, 9 Sherwood Road, Toowong QLD 4066, Australia, Attention: Corporate Secretary. Our Corporate Secretary shall initially review and compile all such communications and may summarize such communications prior to forwarding to the appropriate party. Our Corporate Secretary will not forward communications that are not relevant to the duties and responsibilities of our Board. Our Board will generally respond, or cause us to respond, in writing to bona fide communications from stockholders addressed to one or more members of our Board.
Delinquent Section 16(a) Reports
The rules of the SEC require that the Company disclose late filings of reports of stock ownership (and changes in stock ownership) by its directors, executive officers, and beneficial owners of more than ten percent of the Company’s stock. The Company has undertaken responsibility for preparing and filing the stock ownership forms required under Section 16(a) of the Exchange Act, as amended, on behalf of its officers and directors. Based upon a review of forms filed and information provided by the Company’s officers and directors, we believe that all Section 16(a) reporting requirements were met during fiscal year 2025.
The following is a discussion of compensation arrangements of our NEOs. As an EGC as defined in the JOBS Act, we are not required to include a Compensation Discussion and Analysis section and have elected to comply with the scaled disclosure requirements applicable to emerging growth companies.
Named Executive Officers
The following table sets forth as of January 1, 2026, the names and ages of our executive officers, who also constitute our named NEOs for 2025. Biographies for each executive officer that is not a director, are included below the table. There are no family relationships between the executive officers or between any director and any executive officer.
|
Name
|
Age |
Position
|
||
|
Wayne Paterson
|
59 |
Vice Chairman and Chief Executive Officer
|
||
| Matthew McDonnell | 53 | Chief Financial Officer | ||
|
David St Denis
|
57 |
President, Director (former Chief Operating Officer)
|
Matthew McDonnell
Matthew McDonnell served as ATPL’s Chief Financial Officer from November 2018 and, following the Reorganization, now holds the role of Chief Financial Officer of the Company. He has also served as the Chief Financial Officer of v2vmedtech since September 2023, which we consider to be a VIE and consolidate in our financial statements. Prior to his appointment as Chief Financial Officer of ATPL, Mr. McDonnell worked for KPMG, a global professional services firm, for over 24 years, where he held several senior positions, including 10 years as a partner. He has a broad range of industry experience and corporate governance acumen, having delivered audit, accounting, and advisory services to a broad range of sectors. During his time at KPMG, Mr. McDonnell worked in Australia covering the financial services, transport, industrial markets, health, childcare and energy industries. He has experience in restructurings, acquisitions, divestments, privatizations and other significant financial transactions. Mr. McDonnell also served as a director and Chair of the Audit and Risk Management Committee of the State Library of Queensland for eight years. Mr. McDonnell holds a Bachelor of Economics from Macquarie University, Australia. He is an Associate of Chartered Accountants in Australia and New Zealand, a Fellow of the Financial Services Institute of Australasia and a Member of the Australian Institute of Company Directors.
2025 Summary Compensation Table
The following table and related footnotes provide information concerning the compensation of our NEOs for each of the years ended December 31, 2025 and December 31, 2024.
|
Name and Principal
Position |
Year |
Salary ($)(1) |
Bonus
($) |
Stock awards ($)(2) |
Option awards ($)(2) |
Nonequity incentive plan compensation(3) |
Change in pension value and nonqualified deferred compensation earnings ($) |
All other compensation ($)(4) |
Total ($) | |||||||||||||||||||||||||
|
Wayne Paterson
|
2025
|
734,598 | — | 4,460,000 | — | 896,100 | — | 40,403 | 6,131,101 | |||||||||||||||||||||||||
|
Vice Chairman and Chief Executive Officer
|
2024
|
696,389 | 125,000 | — | 1,461,423 | 725,000 | — | 35,533 | 3,043,345 | |||||||||||||||||||||||||
|
David St Denis
|
2025
|
479,156 | — | — | — | 469,680 | — | 33,069 | 981,905 | |||||||||||||||||||||||||
|
President and Former Chief Operating Officer
|
2024
|
450,000 | 50,000 | 3,000,000 | — | 380,000 | — | 34,596 | 3,914,596 | |||||||||||||||||||||||||
|
Matthew McDonnell
|
2025
|
264,379 | — | — | — | 176,826 | — | 16,147 | 457,352 | |||||||||||||||||||||||||
|
Chief Financial Officer
|
2024
|
267,432 | 50,000 | 499,998 | — | 158,757 | — | 24,471 | 1,000,658 |
(1)
The amounts represent the actual salary paid during the fiscal year. Variances from the prior year are due to (i) timing of base salary adjustments effective December 16, 2024, (ii) movements in annual leave accruals and payouts, and (iii) foreign exchange fluctuations, where applicable. Base salary rates for each NEO did not otherwise change during the fiscal year. The amounts reported in this column for Mr. McDonnell for 2025 and 2024 are presented in USD using exchange rates which averaged over the relevant year to approximately A$1.00 to $0.64 and A$1.00 to $0.65, respectively.
(2)
The amounts reported represent the aggregate grant date fair value for the stock and option awards granted in 2025 and 2024, each computed in accordance with FASB ASC Topic 718. Please refer to note 15 to the company’s consolidated financial statements in this Annual Report on Form 10-K for a description of the assumptions made in the valuation of stock and option awards under FASB ASC Topic 718. For fiscal year 2025, the “Stock Awards” column reflects the grant date fair value of an award (contingently granted on December 16, 2024) of 1,000,000 restricted stock units granted on December 3, 2025 after stockholder approval received on the same date.
(3)
The non-equity incentive plan compensation bonus for Mr. McDonnell, which has been accrued at year-end, has been translated using the spot exchange rate, which was approximately A$1.00 to $0.67.
(4)
The amounts disclosed as “all other compensation” set out above for Messrs. Paterson and St Denis for 2025 reflects amounts related to health and other benefit related payments in the amounts of $25,209 for Mr. Paterson and $25,065 for Mr. St Denis and the Company’s 401(K) match in the amounts of $10,500 for Mr. Paterson and $6,807 for Mr. St Denis. The amount disclosed as “all other compensation” set out above for Mr. McDonnell for 2025 reflects $15,563 of superannuation payments. All such payments to Mr. McDonnell are presented in USD using exchange rates prevailing at the dates of the transactions, which averaged over the year to approximately A$1.00 to $0.66.
Employment Agreements
We have entered into employment agreements with each of our NEOs. The material terms of the employment agreements are described below.
Under the terms of Mr. Paterson’s executive employment agreement, which became effective on December 16, 2024 (the “Paterson Agreement”), Mr. Paterson serves as the Chief Executive Officer of the Company and on our Board as Vice Chairman. Mr. Paterson’s base salary is $725,000, subject to annual review by our Board for increase. Mr. Paterson is also entitled to receive an annual incentive bonus with a target opportunity of up to 100% of his base salary, a long-term incentive compensation award with a total target fair value of $6,000,000 in connection with the Company’s IPO, and, starting with the 2026 calendar year, annual long-term incentive compensation awards, which will be granted in 2026 and will vest based upon future service and/or performance after the grant date, with a total target grant value of $4,000,000 subject to approval by stockholders in accordance with the rules of the ASX. The Paterson Agreement may be terminated by the Company without “cause” or by Mr. Paterson without “good reason” (as each term is defined in the Paterson Agreement) upon six months’ written notice to the other party or by Mr. Paterson for “good reason” by giving notice to the Company of the existence of a “good reason” trigger within sixty days of its occurrence, the Company failing to cure the trigger within the following thirty days, and Mr. Paterson terminating his employment within thirty days of the end of the cure period. If the Company terminates Mr. Paterson’s employment without “cause” or Mr. Paterson terminates employment for “good reason,” Mr. Paterson will receive the following compensation and benefits, subject to his executing and not revoking a release of claims against the Company and its affiliates: the Company will continue to pay Mr. Paterson his base salary for twelve months, Mr. Paterson will be entitled to receive a pro-rata portion of his annual bonus for the year of termination based on actual achievement of the applicable performance metrics, any outstanding equity awards that vest solely based on continuous service will be earned on a pro-rata basis, and any outstanding performance-based equity awards will be earned on a pro-rata basis, subject to the achievement of the applicable performance metrics, and the Company will reimburse Mr. Paterson for COBRA premium payments for twelve months (or until Mr. Paterson becomes eligible for coverage under another medical plan, whichever occurs first). Mr. Paterson will also be entitled to participate in the Company’s employee benefit plans, programs and policies for senior executives. The Paterson Agreement includes customary non-competition, non-solicitation, intellectual property, and confidentiality provisions.
Mr. St Denis’ executive employment agreement, which became effective on December 16, 2024 (the “St Denis Agreement”) provides for Mr. St Denis’ service as the Chief Operations Officer of the Company. Effective March 5, 2025, Mr. St Denis was appointed as our President and a member of our Board. Pursuant to the St Denis Agreement, Mr. St Denis’ base salary is $475,000, subject to annual review by the Company for increase. Mr. St Denis is also entitled to receive an annual incentive bonus with a target opportunity of up to 80% of his base salary, and, starting with the 2026 calendar year, annual long-term incentive compensation awards, which will be granted in 2026 and will vest based upon future service and/or performance after the grant date, with a total target grant value of $2,000,000 subject to approval by stockholders in accordance with the rules of the ASX. The St Denis Agreement also provided for a long-term incentive compensation award with a total target fair value of $3,000,000 in connection with the Company’s IPO. The St Denis Agreement may be terminated by the Company without “cause” or by Mr. St Denis upon three months’ written notice to the other party. If the Company terminates Mr. St Denis’ employment without “cause”, subject to Mr. St Denis executing and not revoking a release of claims against the Company and its affiliates, the Company will continue to pay Mr. St Denis his base salary for nine months and will reimburse Mr. St Denis for COBRA premium payments for nine months (or until Mr. St Denis becomes eligible for coverage under another medical plan, whichever occurs first). Mr. St Denis is also provided a cellphone and is entitled to participate in the Company’s employee benefit plans, programs and policies for senior executives. The St Denis Agreement includes customary non-competition, non- solicitation, intellectual property, and confidentiality provisions.
Under the terms of Mr. McDonnell’s executive employment agreement, which became effective on December 16, 2024 and which supersedes and replaces all prior agreements related to Mr. McDonnell’s employment (the “McDonnell Contract”), Mr. McDonnell serves as the Chief Financial Officer of the Company. Mr. McDonnell’s base salary is $254,334 (A$380,000), converted using the spot exchange rate of approximately A$1.00 to $0.67 on December 31, 2025 and excluding superannuation. Mr. McDonnell is entitled to contributions to defined contribution plans (superannuation) at 12.5% (previously 12.0%) of the annual eligible compensation (post July 1, 2025) subject to certain contribution caps. Mr. McDonnell is also entitled to receive an annual incentive bonus of up to 60% of his base salary, and, starting with the 2026 calendar year, annual long-term incentive compensation awards, which will be granted in 2026 and will vest based upon future service and/or performance after the grant date, with a total target grant value of $500,000. The McDonnell Contract also provided for a long-term incentive compensation award with a total target fair value of $500,000 in connection with the Company’s IPO. Either party may terminate the employment relationship generally upon three months’ written notice to the other party. The McDonnell Contract includes non-competition, non-solicitation, intellectual property, and confidentiality provisions.
Short-Term Incentive Compensation
Compensation for individuals is linked to our performance as well as the performance and contribution of the individual. Incentive payments are dependent on defined corporate and individual key performance targets being met. Incentive payments for the NEOs and for our broader company are at the discretion of our Board and the Compensation Committee.
The Compensation Committee believes the setting of key corporate and individual key performance targets which are aligned to the corporate strategy, will drive the development, performance and position of our company. The Compensation Committee expects that this will drive increased stockholder returns going forward.
2025 STI Bonuses
The NEOs’ 2025 short-term incentive (“STI”) bonus performance targets were based on a percentage of their base salaries with the actual incentive dependent on certain company and individual performance conditions being satisfied. 2025 STI targets were based on a combination of financial,
capital and strategic objectives. These included adjusted EBITDA
(earnings before interest, tax, depreciation and amortization), a capital
position target including capital transaction(s), and achievement of key
strategic objectives. The strategic objectives for 2025 included milestones
related to the approval and enrollment of the Global Pivotal Registration
Trial, as well as share‑price performance.
|
Name
|
|
Principal Position
|
2025 Target STI Bonus %
|
|
|
Wayne Paterson
|
Vice Chairman and Chief Executive Officer
|
|
100% of base salary
|
|
|
David St Denis
|
|
President, Director (former Chief Operating Officer)
|
|
80% of base salary
|
|
Matthew McDonnell
|
|
Chief Financial Officer
|
|
60% of base salary
|
In early 2026, the Compensation Committee reviewed performance against the 2025 STI performance targets and considered the results achieved during the year, including significant operational and strategic progress. In particular, the Committee considered Mr. Paterson’s significant leadership role and achievements in advancing capital‑related matters during 2025 and for the significant contributions of Messrs. Paterson and St Denis in advancing and initiating the Global Pivotal Registration Trial. Based on this assessment, Messrs. Paterson and St Denis were each awarded an STI bonus equal to 120% of their respective target 2025 STI opportunity. Mr. McDonnell was awarded an STI bonus equal to 100% of his target 2025 STI opportunity, in each case payable in March 2026 based on the salary levels in effect at the time of payment.
Policies and Practices Related to the Grant of Certain Equity Awards
The Compensation Committee approves equity awards granted to the NEOs on or before the grant date. The Compensation Committee does not take material nonpublic information into account when determining the timing and terms of such equity awards, which, for our CEO, are also subject to approval by our stockholders in accordance with the requirements of the ASX. We have not timed the disclosure of material nonpublic information for the purpose of affecting the value of executive compensation. In 2025, we did not award any stock options to the NEOs during any period beginning four business days before the filing of a periodic report on Form 10-Q or Form 10-K or the filing or furnishing of a current report on Form 8-K disclosing material non-public information and ending one business day after the filing or furnishing of such report with the SEC.
Executive Equity-Based Compensation
During 2024, the Company granted 300,000 options to Mr. Paterson with an exercise price of $14.64, pursuant to stockholder approval, which vest in three equal installments on the first three anniversaries of June 19, 2024, subject to Mr. Paterson’s continued employment through each vesting date. The above quoted exercise price has been translated from AUD using the spot exchange rate on the date of the Reorganization which was approximately A$1.00 to $0.64.
In connection with our IPO in 2024, priced at $6.00 per share, each NEO received an award of restricted stock units under the Equity Plan (as defined below), which vests annually over a three-year period. Vesting of such awards will accelerate upon a termination of employment following a change in control. The NEOs received the following number of restricted stock units: Mr. Paterson, 1,000,000; Mr. St Denis, 500,000; and Mr. McDonnell, 83,333. Because Mr. Paterson’s restricted stock units were subject to stockholder approval, his award is considered granted for purposes of this disclosure on December 3, 2025 following approval by stockholders. No equity awards were otherwise granted to the NEOs in 2025.
Equity Compensation Plans
The Company has two historic long-term incentive plans from which no further options or awards will be issued, which were assumed by the Company in connection with the Reorganization:
●
the Admedus Ltd (now known as “n/k/a” ATGC) Employee Long Term Incentive Plan (the “2017 Incentive Plan”), which was approved by stockholders in November 2017; and
●
the Employee Incentive Plan (the “2020 Incentive Plan”).
Under each plan, certain eligible participants could receive ordinary shares, options or rights. The vesting of shares, options or rights may be subject to the satisfaction of service-based conditions and performance hurdles which, when satisfied, will allow eligible participants to receive shares or vested options or rights which are exercisable over shares or CDIs.
Anteris Technologies Global Corp. Equity Incentive Plan
The Company has adopted the Anteris Technologies Global Corp. Equity Incentive Plan (the “Equity Plan”) for purposes of granting options in the Company and other awards based on the shares of the Company to employees and other service providers of the Company. The following is a summary of the material terms of the Equity Plan, which is qualified in its entirety by reference to the full text of the Equity Plan, which was filed as exhibit 10.1 to the Company’s Amendment No. 1 to Form S-1, filed with the SEC on December 9, 2024.
Purpose of the Equity Plan
The purpose of the Equity Plan is to permit award grants to non-employee directors, officers and other employees of the Company and its subsidiaries, and certain consultants to the Company and its subsidiaries, and to provide to such persons incentives and rewards for service and/or performance.
Administration of the Equity Plan
The Equity Plan is administered by the Compensation Committee of our Board or a subcommittee thereof (the “Administrator”). The Administrator has the power to interpret and construe the Equity Plan and is authorized to take any action it determines in its sole discretion to be appropriate, subject to the express limitations contained in the Equity Plan.
Number of Authorized Shares
The aggregate number of shares of the Company’s common stock, par value $0.0001 per share (“Common Stock”) which may be issued or transferred pursuant to awards granted under the Equity Plan will not exceed, in the aggregate, 5,163,023 shares of Common Stock (the “Share Limit”). The Share Limit will be increased by 5% of the total number of issued and outstanding shares of Common Stock on a fully-diluted basis on the last day of the preceding fiscal year on the first day of each fiscal year, for a period of ten years commencing in the first fiscal year following the effective date of the Equity Plan. The aggregate number of shares of Common Stock actually issued or transferred by the Company upon the exercise of incentive stock options (if such awards are able to be granted) will not exceed 4,312,777 shares of Common Stock, which limit will increase by 3% of the total number of issued and outstanding shares of Common Stock at the consummation of the IPO on the first day of each fiscal year, for a period of ten years commencing in the first fiscal year following the effective date of the Equity Plan.
The maximum number of shares subject to awards granted on or following the effective date of the Equity Plan during a single calendar year to any non-employee director, taken together with any cash fees paid to such non-employee director during the fiscal year, may not exceed $750,000 in total value (calculating the value of any such awards based on the fair value of such awards as of their approval effectiveness date); provided, that such calendar year limit shall be $1,000,000 for (i) the non-executive chair of our Board and (ii) a new non-employee director during his or her first year of service on our Board.
In the event of certain changes in the capitalization of the Company, the Administrator will adjust the number and kind of shares of Common Stock available for issuance under the Equity Plan and all awards shall be adjusted as the Compensation Committee, in its sole discretion, determines is equitably required. Except as described below, shares subject to an award under the Equity Plan that are cancelled, forfeited, expire, or become unexercisable without having been exercised in full will be available for subsequent awards under the Equity Plan. Shares withheld in payment of the exercise price of an option or withholding taxes related to an award will be returned to the Share Limit for future grants of awards under the Equity Plan and will not reduce the Share Limit.
Any references in the Equity Plan and this summary to “shares of Common Stock” may be read as a reference to CDIs or shares of Common Stock as the context reasonably requires, unless the contrary intention is expressly stated in the Equity Plan.
Eligibility and Participation
Eligibility to participate in the Equity Plan is generally limited to employees, consultants, directors, and officers of the Company or any subsidiary. Grants of awards over CDIs may only be granted to Australian employees of the Company or a subsidiary who are not U.S. taxpayers.
Types of Awards under the Equity Plan
The Equity Plan authorizes the Administrator to grant awards, individually or collectively, to recipients in any of the following forms, subject to such terms, conditions and provisions as the Administrator may determine to be necessary or desirable:
●
Option rights;
●
Appreciation rights;
●
Restricted stock;
●
RSUs;
●
Cash incentive awards;
●
Performance shares;
●
Performance units (“PSUs”); and
●
Other equity-based awards.
Term of Awards
The term of each award will be determined by the Administrator and stated in the award agreement. In the case of option rights and appreciation rights, the term may not exceed ten years from the grant date or such shorter term as may be provided in the award agreement.
Option Rights
Option rights entitle the option holder to purchase shares of Common Stock at a price established by the Administrator. The Administrator will determine the terms of the option rights, including the vesting and other conditions that must be satisfied for the vesting and exercisability of such awards.
Exercise Price
The Administrator will determine the exercise price of each option right at the date of grant, which price may not be less than 100% of the fair market value of the underlying shares on the date of grant. The Equity Plan prohibits the reduction of the exercise price of option rights without stockholder approval, other than in connection with a change in the Company’s capitalization.
Exercise of Option Rights
An option holder may exercise his or her option rights by delivering notice of the number of option rights that are being exercised accompanied by payment in full of the applicable exercise price, in such form and pursuant to such procedures as the Company may designate from time to time, and may consist of any method of payment by the award agreement and the Equity Plan.
Appreciation Rights
Appreciation rights entitle the holder to receive from the Company an amount determined by the Compensation Committee, which will be expressed as a percentage of the excess of the market value per share of Common Stock on the date when an appreciation right is exercised over the base price provided for with respect to the appreciation right at the time of exercise. The Administrator will determine the terms of the appreciation rights, including the vesting and other conditions that must be satisfied for the vesting and exercisability of such awards.
Base Price
The Administrator will determine the base price of each appreciation right at the date of grant, which price may not be less than 100% of the fair market value of the underlying shares on the date of grant. The Equity Plan prohibits the reduction of the base price of appreciation rights without stockholder approval, other than in connection with a change in the Company’s capitalization.
Stock Awards
Stock awards, including restricted stock, RSUs, cash incentive awards, performance shares, PSUs, and other equity-based awards, may be granted under the Equity Plan. These stock awards may be denominated in shares or units payable in shares of Common Stock (e.g., RSUs), and may be settled in cash, shares, or a combination of cash and shares. Dividend equivalents, which represent a right to receive the equivalent value of dividends paid on shares of Common Stock, may be granted in connection with RSUs, performance shares, and PSUs. The Administrator will determine the terms of stock awards, including the vesting and other conditions that must be satisfied for the vesting of such awards.
Tax Withholding
The Administrator may require a recipient to remit and will have the right to deduct or withhold an amount sufficient to satisfy applicable withholding tax requirements with respect to any award granted under the Equity Plan.
Change in Control
The effect, if any, of certain transactions described in the Equity Plan constituting a change in control of the Company on any awards outstanding at the time immediately prior to such change in control may be specifically set forth in the corresponding award agreement, or if no such treatment is specified, then the Administrator will determine the effect of such transaction on any outstanding awards in accordance with the Equity Plan.
Termination and Amendment of the Equity Plan
Our Board may at any time amend the Equity Plan, subject to any required stockholder approval and any required consent from participants to the extent required under the Equity Plan or by applicable law. Our Board may terminate the Equity Plan at any time; provided, however, that such termination will not affect the rights of holders of outstanding awards granted under the Equity Plan or their successors.
Term of Equity Plan
The Equity Plan became effective in connection with the pricing of the IPO and will continue in effect until terminated through a resolution by our Board, provided that the termination of the Equity Plan will not affect awards then outstanding, and the terms and conditions of the Equity Plan shall continue to apply to such awards. No grant will be made under the Equity Plan on or after the tenth anniversary of the effective date of the Equity Plan.
Outstanding Equity Awards at Fiscal Year End
The following table sets forth the outstanding equity awards held by our NEOs as of December 31, 2025:
| Option awards | Stock awards(1) | |||||||||||||
|
Name
|
Number of
securities underlying unexercised options (#) exercisable |
Number of
securities underlying unexercised options (#) unexercisable |
Equity
incentive plan awards: Number of securities underlying unexercised unearned options (#) |
Option
Exercise Price ($) (2) |
Option
Expiration Date |
Number of
shares or units of stock that have not vested (#) |
Market value
of shares or units of stock that have not vested ($) |
|||||||
|
Wayne Paterson
|
14,358 | — | — | 23.56 | 12/31/2027 | — | — | |||||||
| 31,890 | — | — | 3.76 | 5/15/2029 | — | — | ||||||||
| 41,222 | — | — | 6.04 | 6/13/2027 | — | — | ||||||||
| 258,778 | — | — | 8.25 | 6/13/2027 | — | — | ||||||||
| 466,666 | 233,334(3) | — | 15.28 | 9/15/2028 | — | — | ||||||||
| 100,000 | 200,000(4) | — | 14.64 | 6/19/2029 | — | — | ||||||||
| — | — | — | — | — | 666,667(6) | 3,326,668 | ||||||||
|
David St Denis
|
5,430 | — | — | 23.56 | 12/31/2027 | — | — | |||||||
| 60,000 | — | — | 5.65 | 9/23/2026 | — | — | ||||||||
| 200,000 | — | — | 8.25 | 6/13/2027 | — | — | ||||||||
| — | — | — | — | — | 700,000(5) | 384,568 | ||||||||
| — | — | — | — | — | 333,334(6) | 1,663,337 | ||||||||
|
Matthew McDonnell
|
2,001 | — | — | 4.55 | 7/12/2029 | — | — | |||||||
| 60,000 | — | — | 5.94 | 9/23/2026 | — | — | ||||||||
| 50,000 | — | — | 8.67 | 6/13/2027 | — | — | ||||||||
| — | — | — | — | — | 116,668(7) | 16,969 | ||||||||
| — | — | — | — | — | 55,556(6) | 277,224 | ||||||||
(1)
Stock awards include awards granted under the Share Price Performance Plan (“SPP Units”) and restricted stock units. NEOs who hold SPP Units may receive cash post-vesting that is based on positive increases in the price of the Company’s Common Stock from the base price specified at grant date.
(2)
All options held by Mr. McDonnell are issued in AUD. The exercise prices and share price hurdles have been translated using the year-end spot exchange rate as of December 31, 2025, which was approximately A$1.00 to $0.67.
(3)
Options vest in full on September 15, 2026, subject to Mr. Paterson’s continued employment through the vesting date.
(4)
Options vest in substantially equal installments on June 19, 2026 and June 19, 2027, subject to Mr. Paterson’s continued employment through each vesting date.
(5)
SPP Units vest as follows: the first tranche vests upon our share price reaching $38.20, the second tranche vests upon our share price reaching $47.75, and the third tranche vests on September 13, 2026. The first and second tranches vest and become exercisable on the earlier of the achievement of the specified share price hurdles for ten consecutive trading days and the completion of three years of service. If the share price hurdles for the first and second tranche are not achieved, the options vest on September 13, 2026.
(6)
Restricted stock units vest in substantially equal installments on December 16, 2026 and December 16, 2027, subject to the NEO’s continued employment through such vesting dates.
(7)
SPP Units vest on September 13, 2026, subject to Mr. McDonnell’s continued employment through such vesting dates.
Retirement Plan
Australian employees are entitled to contributions to defined contribution plans (superannuation) at 12.5% of the participant’s annual eligible gross salary and wages (post July 1, 2025) subject to certain contribution caps. The rate has increased by 0.5% annually for the past four years. U.S. employees receive 3% of gross income as an employer contribution limited by the eligible compensation threshold.
Clawback Policy
We have adopted a compensation recovery policy that is compliant with the Nasdaq Listing Rules, as required by the Dodd-Frank Act.
Health Benefit Plan
The Company provides a health benefit plan to U.S.-based NEOs.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table sets forth the amounts of securities authorized for issuance under our compensation plans as of December 31, 2025:
| Plan Category |
Number of securities to be
issued upon exercise of outstanding options, warrants and rights (a) |
Weighted-average exercise
price of outstanding options, warrants and rights (b)(2) |
Number of securities
remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a))(c) | |||||||||
|
Equity compensation plans approved by security holders
|
3,527,547 | (1) | $ | 12.31 | — | |||||||
|
Equity compensation plans not approved by security holders
(3)
|
1,029,851 | (4) | $ | 3.63 | 2,610,662 | (5) | ||||||
|
Total
|
4,557,398 | $ | 12.14 | 2,610,662 | ||||||||
(1)
Reflects 29,646 options outstanding under the 2017 Incentive Plan, 676,751 options outstanding under the 2020 Incentive Plan, and 1,909,748 options outstanding which were granted to directors pursuant to ATPL stockholder approval. Such plans and outstanding options were assumed by the Company in 2024. The balance also includes 911,402 RSUs granted to directors and approved by stockholders in December 2025.
(2)
The weighted-average exercise price relates to outstanding options. The Company’s outstanding restricted stock units have no exercise price.
(3)
See “Anteris Technologies Global Corp. Equity Incentive Plan” above for a description of the material features of the Equity Plan.
(4)
Reflects restricted stock units and options outstanding under the Equity Plan.
(5)
Consists of 2,610,662 shares of Common Stock available under the Equity Plan. In general, the aggregate share limit under the Equity Plan will be automatically increased by 5% of the total number of issued and outstanding shares of Common Stock on a fully-diluted basis on the last day of the preceding fiscal year on the first day of each fiscal year, for a period of ten years commencing in the first fiscal year following the effective date of the Equity Plan.
2025 Director Compensation
The following table and related footnotes show the compensation paid to the then-current members of our Board other than Mr. Paterson, who served as our Chief Executive Officer during 2025, and Mr. St Denis, who served as our President during 2025 (such directors, the “Non-Employee Directors”) during the last completed fiscal year. Dr. Gu resigned from the Board effective as of June 5, 2025 and Mr. Denaro resigned from the Board effective December 13, 2025. Mr. Roberts and Mr. Moss were appointed to the Board effective June 7, 2025. Where applicable, the table includes compensation paid to our Non-Employee Directors in their capacities as directors of the Company during the last completed fiscal year.
|
Name
|
Fees earned or paid in cash ($) |
Stock awards ($)(1) |
Option awards ($)(2) |
Nonequity incentive plan compensation ($) |
Change in pension value and nonqualified deferred compensation earnings ($) |
All other compensation(3) |
Total ($) | |||||||||||||||||||||
|
John Seaberg
|
154,500 | 621,662 | — | — | — | 1,152 | 777,314 | |||||||||||||||||||||
|
Stephen Denaro
(4)
|
115,895 | 310,826 | — | — | — | 9,901 | 436,622 | |||||||||||||||||||||
|
Wenyi Gu
|
30,301 | — | — | — | — | 3,665 | 33,966 | |||||||||||||||||||||
|
David Roberts
|
37,227 | 296,871 | — | — | — | — | 334,098 | |||||||||||||||||||||
|
Gregory Moss
|
32,475 | 296,871 | — | — | — | — | 329,346 | |||||||||||||||||||||
(1)
As of December 31, 2025, our Non-Employee Directors held the following outstanding restricted stock unit (RSU) awards: Mr. Seaberg – 111,609; Mr. Roberts – 66,563; and Mr. Moss - 66,563. During 2025, Mr. Seaberg’s 27,777 RSUs vested and were settled into 27,777 shares of Common Stock. Mr. Denaro and Dr. Gu did not have any outstanding restricted stock unit awards as of December 31, 2025.
(2)
As of December 31, 2025, our Non-Employee Directors held the following outstanding option awards: Mr. Seaberg – 80,000 options exercisable at $8.25, 157,500 options exercisable at $15.28, and 75,000 options exercisable at $14.64; Mr. Denaro – 40,000 options exercisable at $8.67, 80,500 options exercisable at $16.06, and 50,000 options exercisable at $15.39; and Dr. Gu – 80,500 options exercisable at $16.06; Neither Mr. Roberts or Mr. Moss had any outstanding stock option awards as of December 31, 2025. The exercise prices of options held by the Australian directors, which are designated in AUD, have been converted using the year-end spot exchange rate as of December 31, 2025, which was approximately A$1.00 to $0.67.
(3)
The amounts in this column are presented in USD using the average exchange rate for the fiscal year ended December 31, 2025, which was approximately A$1.00 to $0.64. All other compensation amounts relate to superannuation entitlements.
(4)
Mr. Denaro resigned from the Board effective December 13, 2025. In accordance with the terms of the Equity Plan, all unvested RSUs granted to Mr. Denaro during 2025 were forfeited upon his resignation. Accordingly, although the grant‑date fair value of these RSUs is reported in the Director Compensation Table above, Mr. Denaro did not ultimately realize any value from these awards.
Non-Employee Director Compensation Policy
We have adopted a Non-Employee Director Compensation Policy (the “Director Compensation Policy”) to provide for the compensation of non-employee directors for service on our Board. Each non-employee director receives an annual cash retainer for their service in each position as denoted below, each of which is payable in twelve equal monthly installments and is prorated for any portion of a month that a non-employee director is not serving in such position on our Board. Non-employee directors’ annual cash retainers are determined within an aggregate directors’ fee pool limit, which is periodically recommended for approval by stockholders and stands at $2 million per annum, unless increased by approval of stockholders. The 2025 annual cash retainers for each applicable position were:
|
Non-Executive Board Chair*
|
$ | 154,500 | ||
|
Board Member
|
$ | 46,350 | ||
|
Lead Independent Director (if appointed)
|
$ | 25,750 | ||
|
Audit Committee Chair**
|
$ | 20,600 | ||
|
Compensation Committee Chair**
|
$ | 15,450 | ||
|
Nominating & Governance Committee Chair**
|
$ | 10,300 | ||
|
Audit Committee Member
|
$ | 10,300 | ||
|
Compensation Committee Member
|
$ | 7,725 | ||
|
Nominating & Governance Committee Member
|
$ | 5,150 |
*
This is the total cash compensation and the Non-Executive Board Chair is not eligible for any additional cash retainers.
**
Annual retainers for Committee chairs are paid in lieu of, not in addition to, annual retainers for Committee members.
Pursuant to the Director Compensation Policy and subject to stockholder approval on a per grant basis in accordance with the rules of the ASX, unless the underlying security which is acquired on exercise of the option or otherwise on satisfaction of right is purchased on-market (as specified in an award agreement), non-employee directors who are elected or appointed to our Board receive an initial grant of restricted stock units with an aggregate grant date fair value of $250,000, which vests in three equal annual installments, subject to each non-employee director’s continued service through such date. Non-employee directors who serve on our Board as of the date of any annual stockholder meeting and will continue to serve on our Board following such annual stockholder meeting receive, subject to approval on a per grant basis in accordance with the rules of the ASX, unless the underlying security which is acquired on exercise of the option or otherwise on satisfaction of right is purchased on-market (as specified in an award agreement), an annual grant of restricted stock units with an aggregate grant date fair value of $125,000, except for the Non-Executive Board Chair, who receives a grant with a target value of $250,000, which vests on the earlier to occur of the first anniversary of the grant date and the date of our next annual stockholder meeting, subject to each non-employee director’s continued service through such date. A non-employee director who has served on our Board for fewer than six months prior to an annual stockholder meeting receives a prorated grant of restricted stock units, which vests on the earlier to occur of the first anniversary of the grant date and the date of our next annual stockholder meeting, subject to such non-employee director’s continued service through such date. A non-employee director elected for the first time to our Board at an annual stockholder meeting will be eligible to receive only an initial grant of restricted stock units.
Item 12
Security Ownership of Certain Beneficial Owners and Management
The following table provides certain information regarding the ownership of our Common Stock (including our CDIs), as of February 25, 2026 by each person or group of affiliated persons known to us to be the beneficial owner of more than 5% of our Common Stock (including our CDIs); each of our NEOs; each of our directors; and all of our executive officers and directors as a group. The table also sets out the names of all persons (to the best of our knowledge) who have disclosed pursuant to the Corporations Act 2001 (Cth) or in filings made with the SEC that they are “substantial shareholders” of our company and carry 5% or more of the voting rights attached to our issued securities.
Unless otherwise indicated in the table or the related notes thereto, the address for each person named in the table is c/o Anteris Technologies Global Corp. 860 Blue Gentian Road, Suite 340, Eagan, Minnesota 55121.
|
Name of Beneficial Owner
|
Amount and Nature of Beneficial Ownership Common Stock (1) |
Percentage (2) | ||||||||
|
Directors and NEOs
|
||||||||||
|
J. Seaberg
|
257,487 | (3) | * | |||||||
|
W. Paterson
|
1,115,975 | (4) | 1.1 | % | ||||||
|
D. Roberts
|
- | * | ||||||||
|
G. Moss
|
- | * | ||||||||
|
D. St Denis
|
366,512 | (5) | * | |||||||
|
M. McDonnell
|
139,778 | (6) | * | |||||||
|
All directors and executive officers as a group (six persons)
|
1,879,752 | 1.9 | % | |||||||
|
5%+ Stockholders
|
||||||||||
|
L1 Capital Pty Ltd
|
13,219,814 | (7) | 13.6 | % | ||||||
|
Medtronic plc
|
15,652,173 | (8) | 16.1 | % |
* Represents beneficial ownership of less than 1% of the outstanding Common Stock.
(1)
Except as otherwise indicated, we believe that the beneficial owners of the Common Stock listed above, based on information furnished by such owners, have sole investment and voting power with respect to such shares, subject to community property laws where applicable. Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities.
(2)
Percentage of ownership is based on 97,232,054 shares of Common Stock issued and outstanding as of February 25, 2026 (including shares of Common Stock represented by CDIs). Shares of Common Stock underlying options, RSUs or warrants exercisable within 60 days of February 25, 2026 are deemed outstanding for purposes of computing the percentage ownership of the person holding such option or RSUs but are not deemed outstanding for purposes of computing the percentage ownership of any other person.
(3)
Reflects 47,487 shares of Common Stock and 210,000 options to acquire 210,000 shares of Common Stock exercisable within 60 days of February 25, 2026.
(4)
Reflects 203,061 shares of Common Stock and 912,914 options to acquire 912,914 shares of Common Stock exercisable within 60 days of February 25, 2026.
(5)
Reflects 101,082 shares of Common Stock and 265,430 options to acquire 265,430 shares of Common Stock exercisable within 60 days of February 25, 2026.
(6)
Reflects 27,777 shares of Common Stock held directly by Mr. McDonnell and 112,001 options to acquire 112,001 shares of Common Stock exercisable within 60 days of February 25, 2026 held by Quadroo Pty Ltd, as Trustee for the McDonnell Family Trust. Mr. McDonnell and his spouse serve as directors of Quadroo Pty Ltd and share voting and investment power over such shares.
(7)
Represents shares of Common Stock beneficially owned by L1 Capital Pty Ltd, as of January 22, 2026, as reported on the Schedule 13G/A filed by L1 Capital Pty Ltd with the SEC on February 5, 2026. The address for L1 Capital Pty Ltd is Level 45, 101 Collins Street, Melbourne, VIC 3000 Australia.
(8)
Represents shares of Common Stock beneficially owned by Medtronic plc (“Medtronic”) and Covidien Group S.à r.l. (“Covidien”), a wholly owned subsidiary of Medtronic, as of January 22, 2026, as reported on the Schedule 13D filed by Medtronic on January 29, 2026. The address for Medtronic is Building 2, Parkmore Business Park West, Galway, Ireland. The address for Covidien is Ground Floor, Espace Monterery, 40 Av Monterey, L-2163 Luxembourg.
Australian Disclosure Requirements
In addition to the Company’s primary listing on Nasdaq, our shares of Common Stock are also quoted in the form of CDIs on the ASX and trade under the ticker symbol “AVR”. As part of our ASX listing, we are required to comply with the various disclosure requirements as set out under the ASX Listing Rules. The following information is intended to comply with the ASX Listing Rules (where that information has not been provided elsewhere in this Form 10-K).
Issued capital
As of January 31, 2026, the Company’s issued equity instruments consisted of:
•
81,410,333 shares of Common Stock quoted held by 26 stockholders of record on Nasdaq (CDI holder not included);
•
15,821,721 CDIs held by CHESS Depositary Nominees Pty Limited (the “Depositary Nominee”) on behalf of 3,510 stockholders, which are quoted on the ASX;
•
5,385,000 unquoted warrants which entitle the holders of those securities, upon vesting of their conversion rights, to be issued shares of Common Stock (including in certain cases in the form of CDIs) held by 59 holders; and
•
2,668,729 unquoted unlisted options which entitle the holders of those securities, upon vesting of their conversion rights, to be issued shares of Common Stock (including in certain cases in the form of CDIs) held by 102 holders; and
•
1,882,553 unquoted restricted stock units (“RSUs”) which entitle the holders of those securities, upon completion of the service period, to be issued shares of Common Stock (including in certain cases in the form of CDIs) held by 147 holders.
As of January 31, 2026, the Company did not have any restricted securities (within the meaning of the ASX Listing Rules) that are on issue or any securities subject to voluntary escrow that are on issue.
Jurisdiction of Incorporation and Restrictions on the Acquisition of Securities
The Company is incorporated in the State of Delaware in the United States of America. As a foreign company registered in Australia, the Company is not subject to Chapters 6, 6A, 6B and 6C of the Corporations Act 2001 (Cth) dealing with the acquisition of its shares (including substantial holdings and takeovers).
Under the Delaware General Corporation Law (the “DGCL”), our Common Stock are generally freely transferable, subject to restrictions that may be imposed by United States federal or state securities laws, by our Charter or Bylaws or by an agreement binding on our stockholders. The Charter and Bylaws do not impose any specific restrictions on the transfer of our Common Stock. Transfers of the Common Stock will be made only on the transfer books or by a transfer agent designated to transfer the Common Stock. Provisions of the DGCL, the Certificate of Incorporation and the Bylaws could make it more difficult to acquire us by means of a tender offer (takeover), a proxy contest or otherwise, or to remove incumbent officers and directors. These provisions could discourage certain types of coercive takeover practices and takeover bids that our Board may consider inadequate and encourage persons seeking to acquire control of our Company to first negotiate with the Board.
Australian Corporate Governance Statement
The Board and our management are committed to achieving and demonstrating the highest standards of corporate governance. We have reviewed our corporate governance practices against the ASX Operating Rules published by the ASX Corporate Governance Council. Our Corporate Governance Statement sets out the ASX Governance Recommendations and the Company’s response as to how and whether we follow those recommendations.
The Corporate Governance Statement reflects our corporate governance practices in place throughout the financial year. A description of the Group’s current corporate governance practices is set out in our Corporate Governance Statement, which can be viewed at https://anteristech.com/investors/corporate-governance.html.
Our most recent Corporate Governance Statement, dated February 26, 2026 and approved by the Board remains accurate as of the date of this Form 10-K. The Corporate Governance Statement is not incorporated by reference herein.
Voting Rights
Except as otherwise required by law, as provided in our Certificate of Incorporation or as provided in resolutions, if any, adopted by the Board with respect to any series of the preferred stock, the holders of our Common Stock (including CDIs) will exclusively possess all voting power. Each holder of shares of Common Stock will be entitled to one vote for each share held by such holder. Our stockholders do not have cumulative voting rights in the election of directors. Accordingly, holders of a majority of the voting shares are able to elect all of the directors.
Holders of CDIs are entitled to receive notice of, and to attend as guests (but not vote at) meetings of stockholders. The Depositary Nominee (or its custodian) is the legal holder of the shares of Common Stock underlying the CDIs. However, as the beneficial owners of the shares of Common Stock underlying the CDIs, holders of CDIs may:
•
direct the Depositary Nominee (or its custodian) how to vote the shares of Common Stock represented by their CDIs by completing the CDI Voting Instruction Form that accompanies the relevant notice of meeting or proxy statement; or
•
appoint themselves (or another person) to be the Depositary Nominee’s proxy with respect to the shares of Common Stock represented by their CDIs for the purposes of attending and voting at the stockholders meeting by completing the CDI Voting Instruction Form that accompanies the relevant notice of meeting or proxy statement.
Alternatively, holders of CDIs may elect to convert their CDIs into Common Stock and vote those shares of Common Stock at a stockholders meeting. Such conversion must be completed prior to the record date fixed by us for determining the entitlement of stockholders to attend and vote at the stockholders meeting.
Warrants and unquoted options convert into a fixed number of shares of Common Stock or CDIs on various dates in accordance with their respective terms. Each warrant and option carries a specified exercise price and expiration date. Options may be cancelled if the applicable vesting conditions are not satisfied. Holders of warrants and unquoted options have no voting rights and are not entitled to vote at our annual or special meetings of stockholders.
RSUs convert into a fixed number of shares of Common Stock or CDIs upon the satisfaction of the applicable vesting conditions. RSUs may be cancelled if the applicable vesting conditions are not satisfied. Holders of RSUs have no voting rights and are not entitled to vote at our annual or special meetings of stockholders.
Twenty Largest Holders as of January 31, 2026
Below is a statement of the 20 largest holders of shares of Common Stock and CDI holders, the number and percentage of shares of Common Stock held by those holders, and the percentage of shares of Common Stock (including that are held as both Common Stock or CDIs), based on the Company’s registers at January 31, 2026.
Common Stock, excluding CDIs
|
Rank
|
Name
|
Shares of
Common
Stock
|
|
Percentage of
Common Stock
Outstanding (1)
|
|
1
|
CEDE & CO
|
63,039,376
|
|
64.8%
|
|
2
|
COVIDIEN GROUP S.À R.L.
|
15,652,173
|
|
16.1%
|
|
3
|
ADAR1 PARTNERS LP
|
710,205
|
|
0.7%
|
|
4
|
LYTTON-KAMBARA FOUNDATION
|
612,244
|
|
0.6%
|
|
5
|
NANTAHALA CAPITAL PARTNERS LIMITED PARTNERSHIP
|
340,297
|
|
0.3%
|
|
6
|
BLACKWELL PARTNERS LLC SERIES A
|
232,663
|
|
0.2%
|
|
7
|
AMEDAN PTY LTD
|
224,280
|
|
0.2%
|
|
8
|
ADAR1 SPV I LP
|
204,081
|
|
0.2%
|
|
9
|
SPEARHEAD INSURANCE SOLUTIONS IDF LLC SERIES ADAR1
|
106,122
|
|
0.1%
|
|
10
|
MLAD HOLDINGS PTY LTD <A/C MLAD SUPER FUND>
|
73,416
|
|
0.1%
|
|
11
|
NCP RFM LP
|
39,284
|
|
0.0%
|
|
12
|
DANIEL WEE KANG CHIEW
|
37,801
|
|
0.0%
|
|
13
|
AGSC CAPITAL PTY LTD <A/C AGSC CAPITAL
INVESTMENT>
|
30,594
|
|
0.0%
|
|
14
|
AMELIA WEE LYNN CHIEW
|
20,650
|
|
0.0%
|
|
15
|
WAYNE GEOFFREY PATERSON
|
20,334
|
|
0.0%
|
|
16
|
LUCY IK CHIW LAU
|
15,446
|
|
0.0%
|
|
17
|
CHEE HIEN CHIEW
|
12,890
|
|
0.0%
|
|
18
|
NIGEL DOUGLAS WILLIAMS
|
10,000
|
|
0.0%
|
|
19
|
BRENT CHRISTOPHER MARRS
|
9,000
|
|
0.0%
|
|
20
|
ASHLEY JAMES COLE
|
8,069
|
|
0.0%
|
|
|
Total
|
81,398,925
|
|
|
|
|
Remaining Common Stock Holders
|
11,408
|
|
|
|
|
Total Common Stock
|
81,410,333
|
|
|
(1) Percentage of ownership is based on 97,232,054 shares of Common Stock issued and outstanding as of January 31, 2026 (including shares of Common Stock represented by CDIs).
CDIs
|
Rank
|
Name
|
Shares
of CDI
|
Percentage
of
Common
Stock
Outstanding (1)
|
|
|
1
|
HSBC CUSTODY NOMINEES
(AUSTRALIA) LIMITED - A/C 2
|
4,743,359
|
4.9%
|
|
|
2
|
CITICORP NOMINEES PTY
LIMITED
|
1,666,217
|
1.7%
|
|
|
3
|
HSBC CUSTODY NOMINEES
(AUSTRALIA) LIMITED
|
589,599
|
0.6%
|
|
|
4
|
MR PATRICK CHEW
|
427,797
|
0.4%
|
|
|
5
|
BNP PARIBAS NOMINEES
PTY LTD <IB AU NOMS RETAILCLIENT>
|
246,779
|
0.3%
|
|
|
6
|
MUTUAL TRUST PTY LTD
|
230,892
|
0.2%
|
|
|
7
|
THROUGH2 INVESTMENTS
PTY LTD <THROUGH2 SUPER FUND A/C>
|
160,000
|
0.2%
|
|
|
8
|
MR DANIEL BERNARD
CLOUGH
|
130,000
|
0.1%
|
|
|
9
|
HSBC CUSTODY NOMINEES
(AUSTRALIA) LIMITED <GSCO CUSTOMERS A/C>
|
128,335
|
0.1%
|
|
|
10
|
MDM LEBY HWANG
|
128,000
|
0.1%
|
|
|
11
|
MR DAVID LAMM
|
107,100
|
0.1%
|
|
|
12
|
BNP PARIBAS NOMS PTY
LTD
|
92,805
|
0.1%
|
|
|
13
|
MR ROGER ANTHONY
PIERCE
|
82,050
|
0.1%
|
|
|
14
|
BUTTONWOOD NOMINEES
PTY LTD
|
80,103
|
0.1%
|
|
|
15
|
JAMBER INVESTMENTS PTY
LIMITED <THE AMBER SCHWARZ FAM A/C>
|
78,288
|
0.1%
|
|
|
16
|
J P MORGAN NOMINEES
AUSTRALIA PTY LIMITED
|
75,227
|
0.1%
|
|
|
17
|
SUPERIOR COATINGS
(AUST) PTY LTD
|
75,000
|
0.1%
|
|
|
18
|
GAVJUL PTY LTD
<MISSCA SUPER FUND A/C>
|
73,000
|
0.1%
|
|
|
19
|
MR JEE HOH CHIEW
|
68,000
|
0.1%
|
|
|
20
|
MR EDWARD JOHN BROOK +
MR EDWARD ALFRED BROOK <EDWARD BROOK SUPER FUND A/C>
|
67,700
|
0.1%
|
|
|
Total
|
9,250,251
|
|||
|
Remaining CDI Holders
|
6,571,470
|
|||
| Total Common Stock held with CDI shares | 15,821,721 |
(1) Percentage of ownership is based on 97,232,054 shares of Common Stock issued and outstanding as of January 31, 2026 (including shares of Common Stock represented by CDIs).
Significant Unquoted Equity Holders (>20%) at January 31, 2026
As of January 31, 2026, no holder owned 20% or more of the warrants, unlisted options, or RSUs, other than holders which acquired securities under an employee incentive scheme.
Distribution of Common Stock and CDI Holders at January 31, 2026
Below is a distribution schedule of the number of holders of Common Stock and CDIs, categorized by the size of their holdings, based on the Company’s registers as of January 31, 2026.
Common Stock
| Range |
Number of Holders of Record |
Shares of Common
Stock |
Percentage of Common Stock Outstanding (1) |
|||||||
| 1 – 1,000 | 3 | 968 | 0.0% | |||||||
| 1,001 – 5,000 | 4 | 10,440 | 0.0% | |||||||
| 5,001 - 10,000 | 3 | 27,069 | 0.0% | |||||||
| 10,001 - 100,000 | 8 | 250,415 | 0.3% | |||||||
| 100,001 and over | 8 | 81,121,441 | 83.4% | |||||||
| Total | 26 | 81,410,333 |
(1) Percentage of ownership is based on 97,232,054 shares of Common Stock issued and outstanding as of January 31, 2026 (including shares of Common Stock represented by CDIs).
CDIs
| Range |
Number of Holders
of Record |
Shares of CDI |
Percentage of
Common Stock Outstanding (1) |
|||||||
|
1 – 1,000
|
2,566 | 708,737 | 0.7% | |||||||
|
1,001 – 5,000
|
649 | 1,541,640 | 1.6% | |||||||
|
5,001 - 10,000
|
128 | 945,109 | 1.0% | |||||||
| 10,001 - 100,000 | 156 | 4,068,157 | 4.2% | |||||||
|
100,001 and over
|
11 | 8,558,078 | 8.8% | |||||||
| Total | 3,510 | 15,821,721 | ||||||||
(1) Percentage of ownership is based on 97,232,054 shares of Common Stock issued and outstanding as of January 31, 2026 (including shares of Common Stock represented by CDIs).
The Number of Holders Holding Less than a Marketable Parcel of Securities
There were 386 stockholders and/or CDI holders holding less than a marketable parcel of shares of Common Stock and/or CDIs (where a “marketable parcel” means a parcel of securities worth at least A$500, pursuant to the ASX Operating Rules) as of January 31, 2026 at $8.84 per share.
Buy-Back of Securities
There is no active on-market buyback of our securities at this time.
Distribution of Warrant Holders at January 31, 2026
Below is a distribution schedule of the number of holders of warrants, categorized by the size of their holdings, based on our records as of January 31, 2026.
|
Range
|
Number of Holders of
Record |
Units
|
Percentage of
warrants
|
|||||||
| 1 – 1,000 | 11 | 5,439 | 0.1% | |||||||
|
1,001 – 5,000
|
20 | 55,497 | 1.0% | |||||||
|
5,001 - 10,000
|
7 | 55,667 | 1.0% | |||||||
|
10,001 -
100,000
|
9 | 264,892 | 4.9% | |||||||
|
100,001 and
over
|
12 | 5,003,505 | 92.9% | |||||||
|
Round
|
0.1% | |||||||||
|
Total
|
59 | 5,385,000 | 100.0% | |||||||
Distribution of Unlisted Options Holders at January 31, 2026
Below is a distribution schedule of the number of holders of unlisted options, categorized by the size of their holdings, based on our records as of January 31, 2026.
| Range | Number of Holders of Record | Units | Percentage of unquoted options | |||||||
| 1 – 1,000 | 65 | 27,570 | 1.0% | |||||||
| 1,001 – 5,000 | 16 | 31,089 | 1.2% | |||||||
| 5,001 - 10,000 | 5 | 40,666 | 1.5% | |||||||
| 10,001 - 100,000 | 10 | 212,725 | 8.0% | |||||||
| 100,001 and over | 6 | 2,356,679 | 88.3% | |||||||
| Total | 102 | 2,668,729 | 100.0% | |||||||
Distribution of RSU Holders at January 31, 2026
Below is a distribution schedule of the number of holders of RSUs, categorized by the size of their holdings, based on our records as of January 31, 2026.
|
Range
|
Number of Holders of
Record |
Units
|
Percentage of
RSUs
|
|||||||
|
1,001 – 5,000
|
120 | 270,150 | 14.4% | |||||||
|
5,001 - 10,000
|
10 | 74,800 | 4.0% | |||||||
|
10,001 -
100,000
|
13 | 314,882 | 16.7% | |||||||
|
100,001 and
over
|
4 | 1,222,721 | 65.0% | |||||||
|
Round
|
-0.1% | |||||||||
|
Total
|
147 | 1,882,553 | 100.0% | |||||||
General Information
Mr. Wayne Paterson is our Secretary.
The address of our registered and principal administrative office in Australia is Toowong Tower, Level 3, Suite 302, 9 Sherwood Road, Toowong QLD 4066, Australia and our telephone number there is +61 1300 550 310.
Registers of securities are held as follows:
•
for CDIs in Australia, at Computershare Investor Services Pty Ltd, Level 1, 200 Mary Street, Brisbane, QLD 4000 Australia, Investor Enquiries 1300 850 505 (within Australia) +61 3 9415 4000 (outside Australia); and
•
for shares of Common Stock in the United States, at Computershare Investor Services, 150 Royall Street, Canton, MA 02021 USA, Tel: +1 (781) 575 3100.
We advise that we have used the cash and assets in a form readily convertible to cash that we had at the time of our admission to the Official List of ASX in a way that is consistent with our business objectives.
Policies and Procedures with Respect to Related Party Transactions
Our Audit and Risk Committee charter requires that the Audit and Risk Committee review and approve or disapprove all related person transactions that are required to be disclosed by Item 404 of Regulation S-K. We review all relationships and transactions reported to us in which we and our directors and executive officers or their immediate family members or any person who is known by us to be the beneficial owner of more than five percent (5%) of our voting stock are participants to determine whether such persons have a direct or indirect material interest. Our Secretary is primarily responsible for the development and implementation of processes and controls to obtain information from the directors and executive officers with respect to related person transactions and for then determining, based on the facts and circumstances, whether our company or a related person has a direct or indirect material interest in the transaction.
We have a written related-party transaction policy that applies to our executive officers, directors, director nominees, holders of more than 5% of any class of our voting securities and any member of the immediate family of, and any entity affiliated with, any of the foregoing persons. Such persons will not be permitted to enter into a related-party transaction with us without the prior consent of our Audit and Risk Committee, or other independent members of our Board in the event it is inappropriate for our Audit and Risk Committee to review such transaction due to a conflict of interest. Any request for us to enter into a transaction with an executive officer, director, director nominee, principal stockholder, or any of their immediate family members or affiliates, in which the amount involved exceeds $120,000 or one percent (1%) of the average of our total assets as of the end of last three completed fiscal years must first be presented to our Audit and Risk Committee for review, consideration and approval. In approving or rejecting any such proposal, our Audit and Risk Committee will consider the relevant facts and circumstances available and deemed relevant to our Audit and Risk Committee, including, but not limited to, the commercial reasonableness of the terms of the transaction and the materiality and character of the related party’s direct or indirect interest in the transaction.
Certain Relationships and Related Transactions
Other than executive compensation arrangements described elsewhere in this Form 10-K (See “Item 11. Executive Compensation”), described below are the transactions and proposed transactions, since the beginning of our fiscal year ended December 31, 2025, in which we were or are to be a participant and in which any related person has or will have a direct or indirect material interest involving the lesser of $120,000 and one percent (1%) of the average of our total assets as of year-end for the last two completed fiscal years. A related person is any executive officer, director, nominee for director, or holder of 5% or more of our Common Stock, or an immediate family member of any of those persons.
Reorganization Transactions
Prior to the consummation of our IPO, we completed the Reorganization pursuant to which we received all of the issued and outstanding shares of ATPL, which was formerly an Australian public company originally registered in Western Australia, Australia and listed on the ASX, pursuant to the Scheme under Part 5.1 of the Corporations Act. Contemporaneously with implementation of the Scheme, ATPL also cancelled all existing options it has on issue in exchange for our company issuing replacement options to acquire Common Stock pursuant to the Option Scheme under Part 5.1 of the Corporations Act. The Scheme was approved by ATPL’s shareholders at a general meeting of shareholders, which was held on December 3, 2024. The Option Scheme was approved by ATPL’s optionholders at a general meeting of optionholders held on the same day. ATPL obtained approval of the Scheme and the Option Scheme by the Supreme Court of Queensland on December 4, 2024. As a result of the Reorganization, ATPL became a wholly owned subsidiary of our company and the shareholders of ATPL immediately prior to the consummation of the IPO became holders of the either one share of Common Stock for every ordinary share of ATPL or one CDI for every one ordinary share of ATPL for each share held as of the record date.
v2vmedtech Contributions
On April 18, 2023, we purchased 30% of the equity capital stock of v2vmedtech, pursuant to a contribution and stock purchase agreement, and concurrently contributed $0.2 million and entered into a series of agreements (collectively, the “v2v Agreements”) with v2vmedtech. v2vmedtech has a license agreement with Columbia University to develop an innovative heart valve repair device utilizing a transcatheter edge-to-edge repair method for a minimally invasive treatment of mitral and tricuspid valve regurgitation, also known as leaky valve.
Under the terms of the v2v Agreements, we agreed to provide certain development services to v2vmedtech in exchange for equity in v2vmedtech. Pursuant to the v2v Agreements, we provide engineering, clinical, regulatory, marketing, and executive management resources, but excluding medical and chief medical officer services, in connection with v2vmedtech’s development of these valve repair devices. We are responsible for developing products and preparing regulatory filings and all costs and expenses incurred by us directly, related to the development of devices constitute development contributions under the v2v Agreements, for which we are solely responsible. These contributions are to be provided over five stages linked to key development and regulatory requirements for the device for transcatheter edge-to-edge repair of the mitral valve.
During the year ended December 31, 2025, we contributed $2.6 million to v2vmedtech to finance v2vmedtech’s operations. The total amount of eligible development contributions and operational contributions paid by us under the v2v Agreements as of December 31, 2025 was $6.2 million. See “Item 1. Business—Collaborations” in Part I of this Form 10-K for more information on the v2v Agreements.
Deed of Cross Guarantee
ATGC and ATPL (ACN 088 221 078) are party to a deed of cross guarantee dated December 20, 2024 (“Deed”). The Deed remained in place throughout the year ended December 31, 2025, and there were no changes to the parties to the Deed during that period. The Company did not rely on the reporting relief available under the Deed for the year ended December 31, 2025. The Deed was entered into in prior periods for Australian statutory reporting purposes; however, the Company did not rely on the reporting relief available under the Deed for the year ended December 31, 2025, and the Deed had no impact on the preparation or presentation of these consolidated financial statements.
Indemnification Agreements
We have entered into agreements to indemnify our directors and executive officers. These agreements require us to indemnify these individuals for certain expenses (including attorneys’ fees), judgments, fines and settlement amounts reasonably incurred by such person in any action or proceeding, including any action by or in our right, on account of any services undertaken by such person on behalf of our company or that person’s status as a member of our Board or executive officer to the maximum extent allowed under Delaware law.
Medtronic Agreements
Purchase Agreement
On January 20, 2026, the Company entered into a stock purchase agreement (the “Purchase Agreement”) with Medtronic, pursuant to which the Company agreed to issue and sell to Medtronic 15,652,173 shares of Common Stock at a purchase price of $5.75 per share in the Medtronic Private Placement. The Purchase Agreement contained customary representations, warranties and agreements by the Company and Medtronic.
Registration Rights Agreement
On January 22, 2026, the Company and Medtronic entered into the Registration Rights Agreement, pursuant to which the Company agreed to file a registration statement (the “Resale Registration Statement”) covering the resale of the Common Stock sold to Medtronic in the Medtronic Private Placement (the “Registrable Securities”) by Medtronic no later than 18 months following the closing of the Medtronic Private Placement. In addition, pursuant to the Registration Rights Agreement, the Company granted Medtronic the right to demand the sale of the Common Stock sold in the Medtronic Private Placement in one underwritten offering. The Company also agreed to be responsible for all fees and expenses incurred in connection with such registration.
In the event (i) the Resale Registration Statement has not been declared effective in the time period specified in the Registration Rights Agreement or (ii) after the Resale Registration Statement has been declared effective by the SEC, sales cannot be made pursuant to the Resale Registration Statement for any reason, subject to certain limited exceptions, then the Company will agree, pursuant to the Registration Rights Agreement, to make payments to Medtronic as liquidated damages in an amount equal to 1.0% of the aggregate amount paid pursuant to the Purchase Agreement by Medtronic for the Registrable Securities then held by Medtronic for each 30-day period or pro rata for any portion thereof during which such event continues. In no event will such liquidated damages exceed 5.0% of the aggregate purchase price of the shares of Common Stock purchased by Medtronic under the Purchase Agreement.
Pursuant to the Registration Rights Agreement, the Company granted Medtronic customary indemnification rights in connection with the Resale Registration Statement. Medtronic also granted the Company customary indemnification rights in connection with the Resale Registration Statement.
Investor Rights Agreement
On January 22, 2026, the Company and Medtronic entered into an investor rights agreement (the “Investor Rights Agreement”), pursuant to which the Company and Medtronic have certain rights and obligations, including:
●
Participation rights: So long as Medtronic beneficially owns at least 75% of the shares of Common Stock purchased pursuant to the Purchase Agreement (the “Minimum Ownership Threshold”), Medtronic will have participation rights with respect to certain future issuances of equity securities by the Company to maintain Medtronic’s percentage ownership interest, subject to customary carve-outs set forth in the Investor Rights Agreement.
●
Transfer restrictions: From the closing date of the Medtronic Private Placement through May 22, 2027, Medtronic will be prohibited from transferring any of the shares of Common Stock purchased pursuant to the Purchase Agreement, subject to certain customary exceptions. In addition, until January 22, 2029, transfers of the shares of Common Stock purchased pursuant to the Purchase Agreement by Medtronic will be subject to, among other things, volume limitations and prohibitions on transfers to activist investors or any person engaged in the business of structural heart therapeutics.
●
Collaboration: The Company and Medtronic agree to engage in non-exclusive good faith discussions regarding potential collaboration opportunities with respect to manufacturing, co-development and co-commercialization of the Company’s products and other matters as may be agreed to between the Company and Medtronic from time to time.
●
Standstill: From the closing date of the Medtronic Private Placement through May 22, 2027 (the “Standstill Period”), Medtronic will be subject to customary standstill provisions (the “Standstill”), pursuant to which, subject to customary exceptions, neither Medtronic nor any of its affiliates may, directly or indirectly, among other things, acquire additional securities of the Company, propose or pursue extraordinary transactions involving the Company, seek to influence or control the Company’s management or board of directors, solicit proxies, initiate stockholder proposals or coordinate with third parties with respect to any of the foregoing. Notwithstanding the foregoing, following the occurrence of certain fundamental change events, Medtronic may make proposals or offers privately to the Company, subject to the terms of the Investor Rights Agreement.
●
Voting agreement: During the Standstill Period, Medtronic will be obligated to cause all shares of Common Stock beneficially owned by Medtronic and its affiliates to be present for quorum purposes and to be voted in accordance with the recommendations of the Board with respect to all proposals submitted to the stockholders of the Company.
●
Board observer: From the closing date of the Medtronic Private Placement until the Observer Termination Date (as defined below), Medtronic will have the right to designate one non-voting board observer, who must be reasonably acceptable to the Board. Medtronic’s right to designate a board observer will terminate upon the earliest of the date on which (the “Observer Termination Date”): (i) Medtronic ceases to meet the Minimum Ownership Threshold; (ii) certain antitrust or competition law issues arise with respect to the board observer’s continued service as such; (iii) Medtronic breaches the Standstill; (iv) Medtronic takes any of the actions prohibited by the Standstill after the Standstill Period; and (v) Medtronic or the board observer materially breaches any clean team agreement or any provision addressing competitively sensitive information of the Company in any confidentiality agreement.
●
Right to negotiate: From the closing date of the Medtronic Private Placement until the date on which the U.S. Food and Drug Administration approves the Company’s Class III device, and provided that Medtronic meets the Minimum Ownership Threshold and Medtronic’s board observer right has not been terminated pursuant to certain circumstances, the Company will notify Medtronic upon receipt of certain acquisition proposals and, subject to applicable law and the fiduciary duties of the Board, is obligated to provide Medtronic with substantially the same information provided to the party that made such acquisition proposal and negotiate in good faith with Medtronic for a specified period of time. Nothing in the Investor Rights Agreement requires the Company or the Board to approve, recommend or consummate any transaction or limits the ability of the Board to take or refrain from taking any action required to comply with its fiduciary duties under applicable law.
Director Independence
Our Board currently consists of five members. Our Board has determined that Mr. Seaberg, Mr. Moss and Mr. Roberts qualify as independent directors in accordance with the Nasdaq Listing Rules. Our Board has further determined that Mr. Seaberg, Mr. Moss and Mr. Roberts qualify as independent directors under Nasdaq Listing Rules applicable to membership on our Audit and Risk Committee. In addition, our Board has determined that each of the members of our Audit and Risk Committee is “financially literate” pursuant to the Nasdaq Listing Rules, and that Mr. Roberts is an “audit committee financial expert,” as defined in applicable SEC rules, because of his individual extensive financial experience. Mr. Paterson and Mr. St Denis are not considered independent by virtue of their positions as Chief Executive Officer and President (former Chief Operating Officer), respectively, of the company.
Fees billed by KPMG (Brisbane, Australia, (PCAOB ID 1020 ) for the fiscal years ended December 31, 2025 and 2024, respectively, are as follows:
|
| Fiscal Year Ended December 31, | |||||||
|
2025
|
2024
| |||||||
|
Audit fees (1)
|
$ | 513,371 | $ | 649,610 | ||||
|
Audit-related fees
|
- | - | ||||||
|
Tax fees
|
- | - | ||||||
|
All other fees
|
- | - | ||||||
|
Total fees
|
$ | 513,371 | $ | 649,610 | ||||
(1)
Audit fees consist of fees for professional services provided primarily in connection with the annual audit of our financial statements, quarterly reviews and other audit services, including services related to SEC registration statements and other documents such as the preparation of comfort letters and consents.
Determination of Independence
In considering the nature of the services provided by our independent registered public accounting firm, the Audit and Risk Committee determined that such services are compatible with the provision of independent audit services. The Audit and Risk Committee discussed these services with our independent registered public accounting firm and our management to determine that they are permitted under the rules and regulations concerning auditor independence.
Additional information concerning the Audit and Risk Committee and its activities can be found in the “Board Committees — Audit and Risk Committee” section of this Form 10-K.
Pre-Approval Policy
According to policies adopted by the Audit and Risk Committee and ratified by our Board, to ensure compliance with the SEC’s rules regarding auditor independence, all audit and non-audit services to be provided by our independent registered public accounting firm must be preapproved by the Audit and Risk Committee. The Audit and Risk Committee has established a general pre-approval policy for certain audit and non-audit services, up to a specified amount for each identified service that may be provided by the independent auditors.
The Audit and Risk Committee approved one hundred percent (100%) of all services provided by KPMG during the year ended December 31, 2025. The Audit and Risk Committee has considered the nature and amount of the fees billed by KPMG and believes that the provision of the services for activities unrelated to the audit is compatible with maintaining KPMG’s independence.
The following documents are filed as part of this report:
1.
Financial Statements
Information in response to this Item is included in Part II, Item 8 of this Form 10-K.
2.
Financial Statement Schedules
All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
3.
Exhibits
The following is a list of exhibits filed or furnished as part of this Form 10-K.
Exhibit Index
| Exhibit | Description | Incorporated by Reference | Filed | ||
| Number | Form | Date | Number | Herewith | |
|
Scheme Implementation Deed, dated August 13, 2024, by and between Anteris Technologies Global Corp. and Anteris Technologies Ltd
|
S-1 | 11/22/2024 | 2.1 | ||
|
Second Amended and Restated Certificate of Incorporation of Anteris Technologies Global Corp.
|
8-K | 12/16/2024 | 3.1 | ||
|
Amended and Restated Bylaws of Anteris Technologies Global Corp.
|
8-K | 12/16/2024 | 3.2 | ||
|
4.1
|
Reference is made to Exhibits 3.1 through 3.2
|
X | |||
|
Description of Registrant’s Securities
|
10-K | X | |||
|
Form of Common Stock Warrant
|
10-Q | 11/12/2025 | 4.2 | ||
|
Form of Confirmation Letter (containing the terms of the CDI Warrants)
|
10-Q |
11/12/2025
|
4.3 | ||
|
Anteris Technologies Global Corp. Equity Incentive Plan
|
S-1 | 11/22/2024 | 10.1 | ||
|
Admedus Ltd Employee Long Term Incentive Plan
|
S-8 | 12/16/2024 | 99.2 | ||
|
Anteris Technologies Ltd Employee Incentive Plan
|
S-8 | 12/16/2024 | 99.3 | ||
|
Form of Indemnification Agreement for Directors and Officers
|
S-1 | 11/22/2024 | 10.2 | ||
|
Development Agreement, dated April 18, 2023, by and between v2vmedtech, inc. and Anteris Technologies Corporation
|
S-1 | 11/22/2024 | 10.3 | ||
|
Supply and Quality Agreement, dated May 15, 2024, by and between Anteris Aus Operations Pty Ltd and Harvey Industries Group Pty Ltd
|
S-1 | 11/22/2024 | 10.9 | ||
|
Second Amended and Restated Supply and License Agreement, dated June 1, 2018, between 4C Medical Technologies, Inc. and Admedus Corporation
|
S-1 | 11/22/2024 | 10.10 | ||
|
Amendment No. 1 to Second Amended and Restated Supply and License Agreement, dated March 5, 2024, between 4C Medical Technologies, Inc. and Anteris Technologies Corporation
|
S-1 | 11/22/2024 | 10.11 | ||
|
Supply and Quality Agreement, dated November 16, 2021 between Anteris Technologies Corporation and Aran Biomedical Teoranta
|
S-1 | 11/22/2024 | 10.12 | ||
|
First Amended and Restated Services Agreement, dated February 21, 2021, by and between NPX Medical, LLC and Anteris Technologies Corporation
|
S-1 | 11/22/2024 | 10.14 | ||
|
Amendment No. 1 to First Amended and Restated Services Agreement, dated March 24, 2024, by and between NPX Medical, LLC and Anteris Technologies Corporation
|
S-1 | 11/22/2024 | 10.15 | ||
|
Master Services Agreement, dated June 1, 2021, by and between Anteris Technologies Corporation and Switchback Medical LLC
|
S-1 | 11/22/2024 | 10.16 | ||
|
First Amended and Restated Master Services Agreement, dated July 28, 2025, by and between Anteris Technologies Corporation and Switchback Medical LLC
|
10-Q | 11/12/2025 | 10.1 | ||
|
Combined Bioinformatics Master Services Agreement, dated September 1, 2021, by and between Anteris Technologies Corporation and Cardiovascular Research Foundation
|
S-1 | 11/22/2024 | 10.18 | ||
|
Lease of Part 26 Harris Road, Malaga, dated February 1, 2009, by and between Giacomel Pty Ltd, Verigen Australia Pty Ltd and Genzyme Corporation
|
S-1 | 11/22/2024 | 10.19(a) | ||
|
Deed of Variation of Lease, dated June 23, 2014, among Giacomel Pty Ltd, Admedus Biomanufacturing Pty Ltd, Genzyme Corporation and Admedus Ltd
|
S-1 | 11/22/2024 | 10.19(b) | ||
|
Deed of Extension and Variation, dated February 19, 2019, by and between Giacomel Pty Ltd, Admedus Biomanufacturing Pty Ltd and Admedus Ltd
|
S-1 | 11/22/2024 | 10.19(c) | ||
|
Deed of Assignment of Lease, dated March 28, 2023 by and between Giacomel Pty Ltd, Admedus Biomanufacturing Pty Ltd, Admedus Regen Pty Ltd and Anteris Technologies Ltd
|
S-1 | 11/22/2024 | 10.19(d) | ||
|
Deed of Variation of Lease, dated June 12, 2023, by and between Giacomel Pty Ltd, Anteris Aus Operations Pty Ltd and Anteris Technologies Ltd
|
S-1 | 11/22/2024 | 10.19(e) | ||
|
Deed of Extension and Variation of Lease, dated February 13, 2024, among Giacomel Pty Ltd, Anteris Aus Operations Pty Ltd and Anteris Technologies Ltd
|
S-1 | 11/22/2024 | 10.19(f) | ||
|
Professional Services Agreement, dated September 3, 2021, between Anteris Technologies Corporation and Christopher Meduri, M.D.
|
S-1 | 11/22/2024 | 10.20(a) | ||
|
Amendment No. 1 to Professional Services Agreement, dated May 1, 2023, between Anteris Technologies Corporation and Christopher Meduri, M.D.
|
S-1 | 11/22/2024 | 10.20(b) | ||
|
Amended and Restated Employment Agreement, dated November 18, 2024, by and between Anteris Technologies Global Corp. and Wayne Paterson
|
S-1 | 11/22/2024 | 10.24 | ||
|
Contract of Employment, dated November 19, 2024, by and between Anteris Technologies Ltd and Matthew McDonnell
|
S-1 | 11/22/2024 | 10.25 | ||
|
Amended and Restated Employment Agreement, dated November 19, 2024, by and between Anteris Technologies Global Corp. and David St Denis
|
S-1 | 11/22/2024 | 10.26 | ||
|
Anteris Technologies Global Corp. Non-Employee Director Compensation Policy
|
S-1 | 11/22/2024 | 10.27 |
| Exhibit | Description | Incorporated by Reference | Filed | ||
| Number | Form | Date | Number | Herewith | |
|
Contribution and Stock Purchase Agreement, dated April 18, 2023, by and among Anteris Technologies Corporation, v2vmedtech, inc., Dr. Vinayak Bapat, Urmi Bapat, Shalaka Bapat, Susheel Kodali, Michael McDonald and Christopher Meduri
|
S-1/A | 12/9/2024 | 10.28 | ||
|
Stock Purchase Agreement, dated January 20, 2026, by and among Anteris Technologies Global Corp. and Covidien Group S.à r.l.
|
8-K | 1/22/2026 | 10.1 | ||
|
Registration Rights Agreement, dated January 22, 2026, by and among Anteris Technologies Global Corp. and Covidien Group S.à r.l.
|
X | ||||
|
Investor Rights Agreement, dated January 22, 2026, by and among Anteris Technologies Global Corp. and Covidien Group S.à r.l.
|
X | ||||
| 10.41 | Master Services Agreement, dated January 12, 2026, by and between Anteris Technologies Corporation and Bright Research Partners, Inc. | X | |||
|
Code of Business Conduct and Ethics
|
10-K | 3/12/2025 | 14.1 | ||
|
Insider Trading and Securities Dealing Policy
|
10-K | 3/12/2025 | 19.1 | ||
|
Subsidiaries of the Registrant
|
X | ||||
|
Consent of Independent Registered Public Accounting Firm for Anteris Technologies Global Corp.
|
X | ||||
|
Consent for Future Market Insights, Inc.
|
X | ||||
|
Power of Attorney (included in the signature page hereto)
|
X | ||||
|
Certification of Principal Executive Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
X | ||||
|
Certification of Principal Financial Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
X | ||||
|
Certification of Principal Executive Officer and Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
X | ||||
|
Compensation Clawback Policy
|
10-K | 3/12/2025 | 97.1 | ||
*
This certification attached as Exhibit 32.1 that accompanies this Form 10-K, is deemed furnished and not filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Form 10-K, irrespective of any general incorporation language contained in such filing.
#
Certain identified information has been excluded from this exhibit pursuant to Rule 601(b)(10) of Regulation S-K because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential.
†
Certain information in this exhibit has been redacted pursuant to Item 601(a)(6) of Regulation S-K.
^
Schedules and exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. Anteris Technologies Global Corp. agrees to furnish supplementally a copy of any omitted schedule or exhibit to the SEC upon request.
+
Management contract or compensatory plan, contract or arrangement.
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Eagan, State of Minnesota, on the 26th day of February, 2026.
|
|
Anteris Technologies Global Corp
|
||
|
|
|
|
|
|
|
By:
|
/s/ Wayne Paterson | |
|
|
|
Name:
|
Wayne Paterson
|
|
|
|
Title:
|
Vice Chairman and Chief Executive Officer
|
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each individual whose signature appears below constitutes and appoints Wayne Paterson and Matthew McDonnell, and each of them, severally, as his or her true and lawful attorneys-in-fact and agents with the power to act, with or without the other, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in his or her capacity as a director or officer or both, as the case may be, of the Company, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that each of said attorneys-in-fact and agents, or his substitute or substitutes may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
| /s/ Wayne Paterson | Vice Chairman and Chief Executive Officer
(Principal Executive Officer) |
February 26, 2026 | ||
| Wayne Paterson | ||||
| /s/ Matthew McDonnell | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | February 26, 2026 | ||
| Matthew McDonnell | ||||
| /s/ John Seaberg | Chairman of the Board of Directors | February 26, 2026 | ||
| John Seaberg | ||||
| /s/ David St Denis | President and Director | February 26, 2026 | ||
| David St Denis | ||||
| /s/ David Roberts | Director | February 26, 2026 | ||
| David Roberts | ||||
| /s/ Gregory Moss | Director | February 26, 2026 | ||
| Gregory Moss |
136